Evaluación de las prestaciones de diferentes bases de datos NoSQL utilizando una función ranking para la identificación de genes.
Benchmark histórico: cómo interpretarlo hoy
Las medidas de esta memoria comparan versiones, hardware, esquemas y clientes concretos. No demuestran que una base sea universalmente más rápida. Titan es una referencia heredada cuya continuidad se encuentra en JanusGraph, y Neo4j, MongoDB y Cassandra han cambiado de forma sustancial desde estas pruebas.
Un benchmark actual debe fijar versiones de servidor y driver, calentar y repetir mediciones, comprobar índices y durabilidad equivalentes, observar CPU, memoria, disco y red, y publicar datos y consultas. La elección comienza por patrones de acceso, consistencia y operación; después se compara la latencia.
En los últimos años aplicaciones web como Facebook o Youtube han revolucionado el mundo de la informática debido a las grandes cantidades de información que gestionan de forma eficiente y rápida. Este hecho, ha propiciado que las empresas encargadas de gestionar estas Bases de datos hayan desarrollado diversas herramientas para responder a las nuevas necesidades, como por ejemplo las Bases de Datos Nosql. Las principales características de estos sistemas es su funcionamiento sin grandes necesidades de recursos y con la capacidad de gestionar una cantidad de información muy elevada (cuanto más información ahí almacenada, mejor es su rendimiento y más eficiente será su gestión).
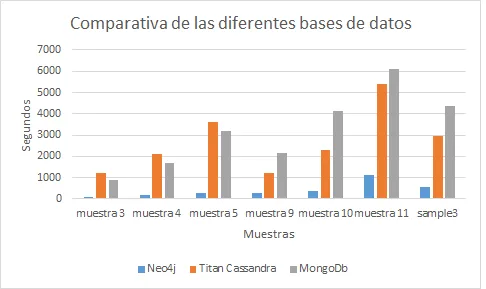
Con el objetivo de hacer una comparativa entre los sistemas más conocidos actualmente en el mercado (Neo4j, Titan Cassandra, MongoDB, Cassandra), hemos decidido aplicar una función Ranking utilizada para analizar genes y ver que sistemas nos ofrecen mejores prestaciones. Para ello, hemos creado diferentes bases de datos de genes con diferente cantidades de conexiones,clases e genes y, de este modo, pode ver cuál sistema realiza los cálculos más rápidamente en diferentes situaciones.
Gracias a esto, hemos llegado a la conclusión que actualmente para sistemas con pocos recursos y no distribuido, obtendremos mejores resultados con aquellas bases de datos NoSql gestionadas por Neo4J, un sistema orientado a grafos y con un lenguaje nativo muy fácil de utilizar.
En esta memoria, se encuentran detalladamente explicados, cuáles han sido las actividades realizadas para la consecución del proyecto y cuáles han sido las herramientas utilizadas.
Palabras clave
Bases de datos, Software libre, NoSql, MongoDB, Cassandra, Titan, Java.
Cómo leer esta comparativa NoSQL
Esta página es larga porque documenta un proyecto académico completo: contexto, modelo de grafos, estrategia de implementación y resultados de rendimiento. La lectura más útil es empezar por la comparación de bases de datos, continuar con la carga de trabajo de ranking sobre grafos e interpretar después los benchmarks según la forma del dato.
Si necesitas una entrada más breve, conviene leerla junto con conceptos básicos y modelos NoSQL, MongoDB actualizado y consultas en Neo4j.
1. Introducción
En el primer capítulo se documentan cual es el contexto, la motivación del alumno, los objetivos propuestos al inicio del proyecto, así como el entorno de trabajo y la estructura de toda la memoria.1.1. Contexto
En la sociedad actual tenemos millones de ejemplos donde manejamos grandes cantidades de información, desde aplicaciones web como Facebook, Twitter, Instagram o bancos con millones y millones de transacciones diarias. En todos estos ejemplos, el manejo veloz de la información y la búsqueda de datos en sus servidores suponen un problema a causa de su gran volumen y, como consecuencia, cualquier operación pasa a ser muy compleja. Para entender mejor la magnitud de este problema existen varias estimaciones donde se afirma por ejemplo que existirá una cantidad de información almacenada y procesada para el año 2020 de 35.000 Exabytes (1 Exabyte son 1024 Petabytes, y un Petabyte 1024 Terabytes), que el tráfico anual de Internet está probablemente cerca de los 8 Exabytes o que el contenido (lo que escribimos, tuiteamos, nuestras fotos, etc) pueden ser aproximadamente unos 500 Exabytes (mmdrigal.com 2014). Como podemos leer, supone un verdadero reto para la informática hasta el punto de crear un nuevo campo para tratar estos problemas. Los investigadores utilizan un nuevo término, “Big Data” para referirse a las situaciones cuando se enfrentan a bases de datos con cantidades gigantescas de información y tienen que solucionar problemas en tres campos muy diferentes entre sí: volumen, velocidad y variedad.- Volumen: las empresas gestionan una cantidad cada vez mayor de datos de todo tipo, acumulando fácilmente terabytes, incluso petabytes, de información.
- Velocidad: a veces 2 minutos es demasiado tarde. En los procesos en los que el tiempo cuenta, como por ejemplo descubrir fraudes, los grandes volúmenes de datos deben utilizarse en tiempo real para maximizar el valor.
- Variedad: los grandes volúmenes de datos incluyen cualquier tipo de datos, estructurados y no estructurados, como texto, datos de sensores, audio, vídeo, secuencias de clics o archivos de registro, entre otros. Al analizar estos datos juntos se encuentra información nueva.
1.2. Motivación
Los algoritmos propuesto para este fin, denominados algoritmos de ranking, utilizan grafos cargados íntegramente en la memoria principal (utilizando estructuras de datos como matrices o listas de adyacencia) para que posteriormente puedan catalogar de los genes no identificados, de acuerdo con el grado de pertenencia a una clase de los genes con los que está relacionado. Estos métodos nos proporcionan como resultado un valor de probabilidad respecto a la relación de pertenencia que existe entre un determinado gen y una clase. Entre todas las diversas técnicas de clasificación encontradas en la bibliografía, en este proyecto, se ha utilizado el Score Function basada en kernels [Re et al, 2010] que se basa en identificar las características de un gen utilizando como base de la información relativa a los genes vecinos, es decir, los vértices que se pueden alcanzar a través de los arcos que sobresalen desde el vértice de inicio. En los nuevos contextos que surgen para aplicar estos métodos, tales como la predicción de características genéticas en diferentes especies o la asociación gen-enfermedad, el número de genes que deben tomarse en consideración, y en consecuencia el tamaño del grafo con el que trabajamos, crece exponencialmente. Por esta razón, los algoritmos de ranking clásicos se vuelven computacionalmente muy costosos o incluso inaplicables y, por lo tanto, tenemos que desarrollar nuevos algoritmos que le permiten escalar mejor la información manteniéndola en un almacenamiento secundario o distribuyendo la carga de trabajo entre varias máquinas. Además, si analizamos los lenguajes actuales o los ordenadores en los que podemos aplicar los algoritmos, solo pueden manejar una matriz de tamaño limitado y rara vez excederá de 50.000 elementos, es decir, como máximo 2.500.000.000 valores para mantener en la memoria. Otra característica de estas matrices es que por su naturaleza se encuentran dispersos y es difícil que un elemento tenga arcos con todos los otros elementos del grafo. Por lo tanto, esto lleva a que la matriz memorice una gran cantidad de valores iguales a 0 que no resultan útiles para el cálculo final. Todas estas nuevas necesidades han propiciado un aumento en la investigación relacionadas con el campo de la gestión de la información masiva, es decir, cada vez más se intentaba solucionar el problema del análisis de la “Big Data”. Como respuesta a este problema en 2009 nació el movimiento NoSQL, un conjunto de organizaciones que proponen sistemas innovadores para la gestión de conjuntos de datos mucho más grandes que las administradas comúnmente a través de los sistemas relacionales. También proponen técnicas eficaces para trabajar con grandes estructuras de grafos, y distribuir la carga de trabajo entre varias máquinas de una forma sencilla e intuitiva. La base de algunos sistemas NoSQL es la posibilidad de utilizar el paradigma MapReduce [Ghemawat et al, 2004], implementado en la biblioteca Hadoop. Estás técnica consta de dividir el problema en una función de Map y una otra de Reduce, de modo que la función de Map convierte las líneas leídas desde el archivo de entrada en pares clave-valor y la lógica interna procese esta pareja, reúna todos los valores de clave y ofrezca el resultado a la función Reduce que procesa el todo. Entre los sistemas que son parte del movimiento NoSQL también incluimos los que manejan estructuras modeladas con un grafo, como Neo4J o Titan Cassandra, es decir, fácilmente representado con nodos y asociaciones entre estos nodos. Esta representación es útil en muchos casos modernos de aplicación (por ejemplo, Facebook) ya que ambos sistemas de gestión de datos abandonan las relaciones de propiedad en favor de una estructura basada en un grafo. Otros sistemas que forman parte de los sistemas NoSQL son, por ejemplo, MongoDB donde guardamos las estructuras de datos en documentos tipo JSON con un esquema dinámico (MongoDB llama ese formato BSON), haciendo que la integración de los datos en ciertas aplicaciones sea más fácil y rápida; o Cassandra, una base de datos NoSQL distribuida, basada en un modelo de almacenamiento en columnas y escrita en Java que posee como característica principal una gran capacidad para gestionar grandes volúmenes de datos en forma distribuida.1.3. Objetivos
Volviendo al problema principal, en este proyecto final de carrera vamos a realizar una búsqueda de información sobre el "Big Data", aplicar los conocimientos adquiridos para desarrollar técnicas para resolver el problema de la predicción de las características genéticas cuando el tamaño de los grafos que se gestionan es de dimensiones elevadas. Siguiendo el espíritu de las bases de datos NoSQL vamos a utilizar máquinas con rendimientos normales (ordenadores personales que podemos encontrar en las tiendas y, por lo tanto, una cantidad relativamente pequeña de la memoria principal) para verificar en qué medida es posible analizar las redes de genes con el fin de predicción de las características genéticas en ellos. El objetivo final es desarrollar nuevos enfoques que serán escalables en el contexto aplicado de referencia. También, queremos evaluar de forma empírica la complejidad de los enfoques desarrollados con el fin de permitir la aplicación de algoritmos de clasificación para grafos de grandes dimensiones. Por lo tanto, para obtener estos resultados es necesario repensar los algoritmos desarrollados para una representación de la matriz de grafos en algoritmos que trabajan directamente en el grafo. Además, es necesario desarrollar algoritmos que trabajen a nivel local en un nodo, en lugar de en forma integral. Esto permite obtener técnicas que son más escalables y la capacidad para paralelizar el proceso de cálculo. Con el fin de alcanzar los objetivos marcados en la tesis hemos decidido implementar los diferentes algoritmos con los siguientes sistemas NoSQL: Neo4J, Titan Cassandra, Cassandra y MongoDB. Cada uno proporcionará a los métodos tienen una filosofía diferente ya que sus especificaciones son bastante diversas entre ellos. El primero de ellos, Neo4J es un sistema de gestión de bases de datos orientado totalmente al modelo de grafos, Titan Cassandra permite la gestión de los grafos pero usando una representación en columnas orientada a esta finalidad, Cassandra representa los grafos a través de la gestión de columnas y por último MongoDB utilizara la gestión de documentos estructurados, que puede ser utilizado para representar grafos. Todos estos sistema permiten la gestión en una sola máquina o en una red. A partir del enfoque proporcionado por los métodos de predicción de las características y funciones genéticas basado en las funciones basada en kernels [King et al, 2012], que es especialmente rápido en memoria principal, hemos realizado diferentes implementaciones en los sistemas NoSQL anteriormente explicados. Después de eso hemos construido diferentes benchmarks para evaluar y comparar el rendimiento de las implementaciones en una sola máquina. Para verificar la exactitud de los resultados proporcionados en las pruebas hemos comparado los valores obtenidos en nuestras implementaciones con los resultados obtenidos en una implementación en R proporcionada por el Profesor Marco Mesiti. Por último, todas las aplicaciones que vamos a implementar se van a caracterizar por los los siguientes requisitos:- Rendimiento: Utilizaremos las estructuras que generen un mejor coste temporal a las aplicaciones. Por ejemplo si queremos comprobar si un elemento está dentro de una colección, el coste temporal de realizar esta consulta a un HashMap es mucho menos que un Array.
- Estabilidad: La aplicación ha sido diseñada para gestionar los errores que ocurran y no se finalice de manera inesperada.
- Portabilidad: Nuestros clientes son programados en java, un lenguaje multiplataforma.
1.4. Descripción del entorno general del proyecto
El proyecto ha sido desarrollado en dos ambientes diferentes. Los 4 primeros meses en un sitio acondicionado en el laboratorio de Bases de datos de la UniversitÀ degli Studi di Milano como parte de un intercambio Erasmus y el resto del curso ha ido alternando entre el domicilio del alumno y algunos laboratorios docentes de la Universidade Positivo de Curitiba (Brasil). Para poder comprobar que el proyecto se desarrollaba correctamente se estableció una serie de entrevistas semanales en las cuales se exponía el trabajo realizado al tutor, además de plantearle las posibles dudas o de discutir algunas decisiones de implementación. Cuando se acabó la beca Erasmus y empezó la de intercambio con Curitiba las reuniones semanales se realizaban por Skype o correo electrónico. El equipo en el que se ha desarrollado todo el proyecto ha sido un Asus UX31 con un procesador Intel Core I 5 (2,2 GHz) con 4Gb de memoria RAM. El sistema operativo utilizado ha sido Windows 7 y las herramientas de desarrollo son software libre. Más adelante en el capítulo [5] se dará más detalles acerca del entorno de desarrollo.1.5. Estructura de documento
El presente documento refleja el proceso de desarrollo del proyecto.Comenzamos el proyecto exponiendo las conclusiones que hemos realizado en las investigaciones bibliográficas iniciales sobre bases de datos NoSQL. En el primer apartado, presentado como Investigación NoSQL ,se presenta las conclusiones obtenidas acerca del problema del Big Data para la gestión de base de datos. Está dividido en diferentes subtareas principales y describe los cuatro sistemas de bases de datos utilizados: Neo4J, MongoDB, Titan Cassandra, Cassandra. El segundo apartado presentamos los métodos de ranking analizados de la bibliografía, que algoritmos utilizan cada uno y define al detalle la función Score basada en kernels aplicada en el proyecto. A continuación, se expone el concepto de validación cruzada para evaluar la calidad del ranking. En el tercer apartado, con toda la investigación inicial terminada, se explica la planificación del proyecto, definiendo las tareas, su duración y fechas estimadas de finalización. En el cuarto apartado vamos a presentar las herramientas utilizadas para realizar este proyecto y el porqué de esta elección. El quinto apartado contiene como hemos construido los diferentes gestores de base de datos y como hemos aplicado el Average Score en cada uno de ellos. Empezaremos explicando en general el algoritmo del Average Score y la validación cruzada, para después especificar un poco más las diferencias de implementación entre Neo4J,Titan Cassandra, MongoDB i Cassandra. El quinto capítulo explica dos subtareas muy importantes. La primera expondremos como hemos verificado los resultados obtenidos por las funciones de Average Score por cada uno de los sistemas y en la segunda explicaremos cuánto tiempo ha sido necesario para realizar los cálculos para diferentes circunstancias. Finalmente se expondrán las conclusión finales del trabajo en el último capítulo2. Planificación del proyecto
En este apartado vamos a comentar las distintas tareas que se han llevado a cabo para la realización del proyecto, así como un diagrama de Gantt donde se puede apreciar tanto la duración temporal de cada una de ellas, como las distintas procedencias que existen entre las tareas.2.1. Definición de las tareas
A continuación se describen cuáles han sido las tareas necesarias para la consecución del proyecto. El proyecto se puede dividir en cinco grandes bloques: definición de los requisitos , aprendizaje sobre las bases de datos Nosql,aprendizaje sobre las funciones de predicción genética, implementación del proyecto y resultados obtenidos en diferentes ejemplos.- Definición de requisitos del sistema Esta tarea está dividida en dos subtareas y su finalidad es obtener los requisitos del proyecto.
- Investigación de los requisitos iniciales Pequeñas entrevistas con el tutor para dejar claro las funcionalidades que debe cubrir el proyecto
- Definición de los requisitos funcionales Realizar una definición de requisitos funcionales con los datos obtenidos en la reunión anterior.
- Investigación sobre el BigData y el movimiento NoSql Esta tarea está dividida en cuatro subtareas y su finalidad es entender que es el problema del Big Data y comprender cómo se desarrollan aplicaciones para resolver este problema mediante las bases de datos NoSql.
- El Big Data y el movimiento NoSql Realizar una búsqueda de información sobre el Big Data y analizar las características del movimiento NoSql
- Familiarizarse con Neo4j Aprendizaje de los conceptos básicos sobre Neo4j,sus características y la realización de un pequeño algoritmo para conocer su funcionamiento.
- Familiarizarse con Titan Cassandra Aprendizaje de los conceptos básicos sobre Titan-Cassandra,sus características y la realización de un pequeño algoritmo para conocer su funcionamiento.
- Familiarizarse con MongoDb Aprendizaje de los conceptos básicos sobre MongoDb,sus características y la realización de un pequeño algoritmo para conocer su funcionamiento.
- Familiarizarse con Cassandra Aprendizaje de los conceptos básicos sobre Cassandra,sus características y la realización de un pequeño algoritmo para conocer su funcionamiento.
- Funciones de predicción genética Esta tarea está dividida en cuatro subtareas y su finalidad es entender que es el problema del Big Data y comprender cómo se desarrollan aplicaciones para resolver este problema mediante las bases de datos NoSql.
- Los métodos utilizados en la lectura Analizamos si es posible representar una red de genes en un grafo, documento o columnas Posteriormente,analizaremos si es posible aplicar los métodos de identificación de genes sobre estas estructuras.
- Función de ranking basada en kernels Recopilación de información sobre este método.
- Técnicas de validación cruzada Recopilación de información sobre este método para la aplicación en las funciones Ranking.
- Análisis y desarrollo de las diferentes bases de datos utilizadas en el proyecto Esta tarea la componen de 7 subtareas cuya finalidad es realizar diferentes programas que realicen los cálculos matemáticos exigidos por los requisitos y comparar la eficiencia de esta operación.
- Analizar y Diseñar la función Score basada en Kernels En esta tarea vamos a diseñar el algoritmo para un cliente de la base de datos. De esta forma vamos a definir los consultas y la metodología a utilizar posteriormente por cada cada base de datos.
- Implementación de la base de datos en Neo4j Analizar qué clases deben ser creadas para formar parte del sistema y estudiar el diseño siguiendo los principios de rendimiento, usabilidad, disponibilidad, escalabilidad, interoperabilidad estabilidad, operatividad y coste. Programas en java los algoritmos correspondiente y compruebas la igualdad entre los resultados obtenidos y los teóricos.
- Implementación de la base de datos en Titan Cassandra Analizar qué clases deben ser creadas para formar parte del sistema y estudiar el diseño siguiendo los principios de rendimiento, usabilidad, disponibilidad, escalabilidad, interoperabilidad estabilidad, operatividad y coste. Programas en java los algoritmos correspondiente y compruebas la igualdad entre los resultados obtenidos y los teóricos.
- Implementación de la base de datos en MongoDb Analizar qué clases deben ser creadas para formar parte del sistema y estudiar el diseño siguiendo los principios de rendimiento, usabilidad, disponibilidad, escalabilidad, interoperabilidad estabilidad, operatividad y coste. Programas en java los algoritmos correspondiente y compruebas la igualdad entre los resultados obtenidos y los teóricos.
- Implementación de la base de datos en Cassandra Analizar qué clases deben ser creadas para formar parte del sistema y estudiar el diseño siguiendo los principios de rendimiento, usabilidad, disponibilidad, escalabilidad, interoperabilidad estabilidad, operatividad y coste. Programas en java los algoritmos correspondiente y compruebas la igualdad entre los resultados obtenidos y los teóricos.
- Verificar Resultados Analizar los resultados obtenidos en cada una de las implementaciones y comprobar que son los deseados.
- Resultados Esta tarea del proyecto consiste en aplicar todos los programas implementados sobre diferentes bases de datos con diversas para analizar los sistemas con mejores prestaciones en cada circunstancia.
- Conclusiones Tarea para establecer las conclusiones a partir de los resultados obtenidos.
- Documentación del proyecto Finalmente se puede considerar también como tarea la redacción de la memoria del proyecto. En esta se puede ver reflejado como ha ido evolucionando el proyecto.
2.2. Diagrama de Gantt
El siguiente diagrama de Gantt (figura [2] ) permite visualizar las duraciones planificadas, fecha de inicio y finalización, y estructura de precedencias al inicio del desarrollo del proyecto. El horario de trabajo contemplado en el diagrama es: lunes a viernes de 9:00 a 13:00 y 15:00 a 16:30.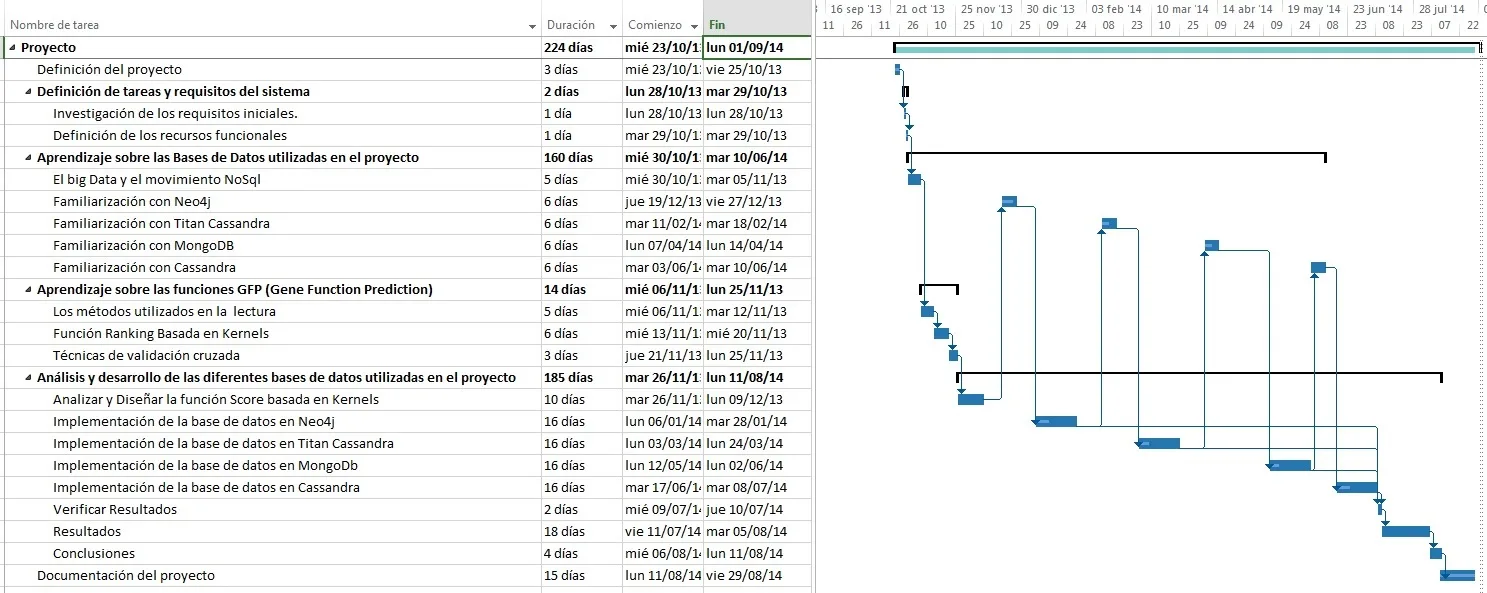
3.Investigación sobre el Big Data y el movimiento NoSQL
Cada día la humanidad produce una cantidad cada vez mayor de información, byte . Baste decir que el 90% de la información creada en las últimas décadas ha sido producida estos dos últimos años. ¿De dónde obtiene toda esta información? Prácticamente en todas partes; en este mundo tecnológico actual, se puede obtener información de cualquier cosa. Ya sea que estemos hablando de una red de sensores, de nuestra última foto subida al Instagram o del último mensaje escrito en Facebook. En este contexto, surge el concepto de Big Data, es decir, el término que utilizan los investigadores para representan el conjunto de información que una organización quiere analizar, pero debido a su tamaño, analizarla resulta muy complejo. El capítulo está organizado de la siguiente manera. En la sección [3.1] se introduce un poco a la historia y concepto de Big Data. Explicamos como ha evolucionado el problema y como el movimiento NoSQL (Not Only SQL) propuso una solución, desarrollando nuevas alternativas a las DBMS relacionales que existían hasta el momento. Entre estos nuevos sistemas de gestión de bases de datos, hemos explicado los conceptos básicos de: Neo4J en la sección [3.2] , Titan Cassandra en la sección [3.3] , MongoDB en la sección [3.4] y Cassandra en la sección [3.5] . Todos estos sistemas se caracterizan por permitir gestionar información organizada en grafos, por lo tanto, permiten aplicar los métodos de predicción de las características genéticas para cumplir con los objetivos del punto 1.3.3.1. El Big Data y el movimiento NoSQL
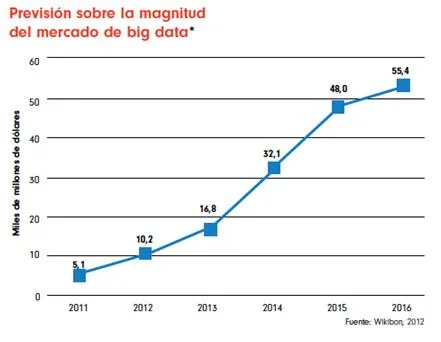
Si analizamos la historia reciente de la humanidad, en 1995 la cantidad de personas en el mundo que tenían acceso a Internet era prácticamente inexistente, pero en estos últimos 10 años la situación ha dado un giro debido a una eclosión de la red y un aumento exponencial de usuarios con nuevas necesidades. Según los datos en diciembre de 2010 el 30% de la población mundial, aproximadamente 2.000 millones de personas, tiene un acceso a Internet y, las proyecciones indican que este ascenso trepidante se disparará en muy pocos años debido a la irrupción de la Internet móvil y los Smartphones (actualmente se estima una cantidad de 1.038 millones de Smartphone con acceso a Internet en el mundo). Así, la cantidad de información publicada, visualizada o subida por los internautas en las aplicaciones más populares de la actualidad alcanza cifras simplemente impresionantes. Es más, se prevé que en los próximos años las cantidades actuales de información que estamos utilizando serán ridículas como podemos observar en la Figura [3.1] . En la primera parte del gráfico se puede ver un círculo donde compara la cantidad de información creada en el 2005 con la información que se prevé crear en 2020. El segundo gráfico nos muestra una evolución de la cantidad global de información y también la capacidad existente de almacenamiento disponible a nivel mundial. Del año 2010 al 2012 es una estimación y del 2012 al 2020 pronósticos.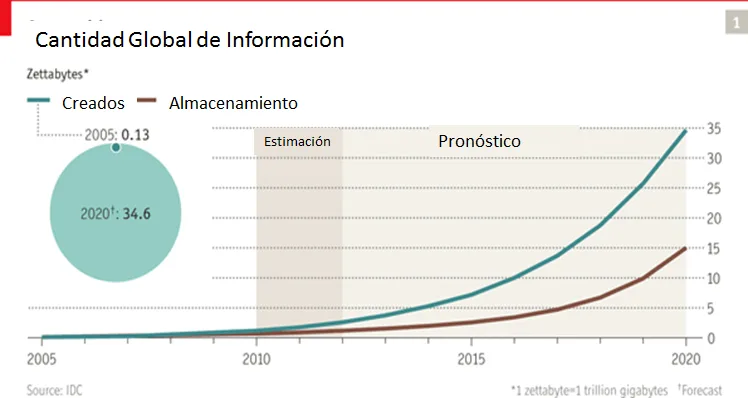
- Youtube: Cada minuto del día se suben unas 24 horas de vídeo a Youtube.
- Twitter: 65 millones de Tweets son compartidos por los internautas en esta red de microblogging; es decir, unos 45.000 Tweets por minuto.
- Facebook: La red social más popular de todas y en todo el mundo ya ha sobrepasado los 500 millones de usuarios registrados, que de media producen mensualmente unas 90 unidades de contenido cada uno, lo que da un total de 45 mil millones de publicaciones mensuales.
- Wikipedia: La popular enciclopedia online y que sentó las bases de la Web 2.0 al presentar un esquema colaborativo y de publicación abierta no ha sido inmune a este crecimiento exponencial, ya que si en 2001 tenía 19.700 páginas publicadas, en la actualidad esta cifra se estima en más de 3.5 millones.
- Volumen. El volumen de los datos almacenados en los depósitos de las empresas ha pasado de ocupar megabytes y gigabytes a petabytes, es decir, el tamaño físico de esta información es a menudo tan grande que es difícil de manejar por la mayoría de las bases de datos. Podemos tomar de ejemplo la Bolsa de Nueva York, que genera un terabyte de datos al día o Twitter, que genera 8 terabytes al día (o 80 MB por segundo).
- Velocidad: La información debe ser procesada velozmente, a menudo en tiempo real. Por ejemplo, eBay se enfrenta al fraude a través de PayPal analizando cinco millones de transacciones en tiempo real al día.
- Variedad: La variedad de datos ha explotado, pasando de ser datos almacenados y estructurados, guardados en bases de datos relacionales, a ser totalmente desestructurados y contener por ejemplo audio, video, XML, texto, etc.
3.1.1 RDBMS VS NoSQL
Hoy en día, cualquier proceso industrial requiere de procesamiento de grandes cantidades de datos y con ese fin comenzaron a florecer paradigmas y algoritmos orientados a este tipo de procesamiento. El análisis de Big Data generalmente incluye todos los datos que tiene en su poder una empresa y ofrece varios beneficios como conducir a nuevos procesos de toma de decisiones, nuevas ideas de marketing o nuevos desarrollos productivos. En esencia, permite a la empresa mantener el ritmo de los tiempos y decidir su futuro. Algunos procesos de negocio son capaces de producir más de 1 TB de datos cada día, por lo que deben encontrar una manera de poner en paralelo la ejecución y mejorar los tiempos de consulta. Primero, como respuesta a estas nuevas necesidades, utilizaron los sistemas de gestión de datos relacionales ( RDBMS) -- Relational Database Management System) que, con las implementaciones de DB2 y Oracle, por su manera sencilla e intuitiva para representar la información en forma de tabla. El éxito del sistema relacional se debió principalmente a la capacidad de manejar las transacciones de operaciones que cumplen con las propiedades ACID:- Atomicidad: es la propiedad que asegura que un operación nunca se podrá quedar a medias ante un fallo del sistema. Entonces podremos considerar que una operación es atómica cuando es imposible para otra parte de un sistema encontrar pasos intermedios. Por ejemplo, en el caso de una transacción bancaria o se ejecuta tanto el depósito y la deducción o ninguna acción es realizada.
- Consistencia: Es la propiedad que asegura que sólo se empieza aquello que se puede acabar. Como consecuencia, se ejecutarán solamente aquellas operaciones que no van a romper las reglas y directrices de integridad de la base de datos.
- Aislamiento: es la propiedad que asegura que una operación no puede afectar a otras. Esto permite que la realización de dos transacciones sobre la misma información sean independientes y no generen ningún tipo de error. Esta propiedad define cómo y cuándo los cambios producidos por una operación se hacen visibles para las demás operaciones concurrentes.
- Durabilidad: Es la propiedad que asegura que una vez realizada la operación, ésta persistirá y no se podrá deshacer aunque falle el sistema y que de esta forma los datos sobreviven de alguna manera.
- no siguen un modelo relacional
- no usan SQL como principal lenguaje de consulta
- poseen la capacidad de escalabilidad horizontales
- utiliza eficientemente los índices y la RAM
- dispone de la capacidad de agregar de forma dinámica nuevos registros y atributos
- no siguen la propiedad ACID.
- Strong ( C)onsistency: todos los clientes deben ver la misma versión de los datos.
- High ( A)vailability: un cliente siempre debe tener disponible al menos una copia de los datos, incluso en presencia de fallos.
- ( P)artition-tolerance: las características del sistema no se deben degradar, incluso si el cálculo se divide en múltiples servidores.
- Consistencia Eventual: No se implementan mecanismos rígidos de consistencia como los presentes en las bases de datos relacionales, donde la confirmación de un cambio implica una comunicación del mismo a todos los nodos que lo repliquen. Poseen una estructura distribuida donde generalmente se distribuyen los datos mediante mecanismos de tablas de hash distribuidas.
- Escalabilidad horizontal: El sistema podrá aumentar el rendimiento del sistema simplemente añadiendo más nodos, sin necesidad en muchos casos de realizar ninguna otra operación más que indicar al sistema cuáles son los nodos disponibles. Muchos sistemas NoSQL permiten utilizar consultas del tipo Map-Reduce, las cuales pueden ejecutarse en todos los nodos a la vez (cada uno operando sobre una porción de los datos) y reunir luego los resultados antes de devolverlos.
- Tolerancia a fallos y Redundancia.
- No generan cuellos de botella: el problema de fondo de los sistemas SQL, es que deben de transcribir cada sentencia para poder ser ejecutada y, cada sentencia compleja requiere, además de un nivel de ejecución más concreto para poderse llevar a cabo, por lo que constituye un punto de entrada común, único y conflictivo en base de rendimiento.
- Solo lo necesario: son sistemas simples que no tienen un sistema de consulta complejo ni con capacidad declarativa para en una sola línea realizar una cantidad interna de operaciones desorbitada.
- Estructura dinámica: Los datos no tienen una definición de atributos fija, es decir: Cada registro puede contener una información con diferente forma cada vez, pudiendo así almacenar sólo los atributos que interesen en cada uno de ellos, facilitando el polimorfismo de datos bajo una misma colección de información. También se pueden almacenar estructuras de datos complejas en un sólo documento, como por ejemplo almacenar la información sobre una publicación de un blog (título, cuerpo de texto, autor, etc) junto a los comentarios y etiquetas vertidos sobre el mismo, todo en un único registro. Hacerlo así aumenta la claridad (al tener todos los datos relacionados en un mismo bloque de información) y el rendimiento (no hay que hacer un JOIN para obtener los datos relacionados, pues éstos se encuentran directamente en el mismo documento).
3.1.2 Tipos de bases de datos NoSQL
Los tipos de bases de datos NoSQL que podemos encontrar son:- Key-Value: clave-valor es la forma más típica, como un HashMap donde cada elemento está identificado por una llave única, lo que permite la recuperación de la información de manera muy rápida. Normalmente el valor se almacenar como un objeto llamado blob (Binary Large Objects) que son elementos utilizados en las bases de datos para almacenar datos de gran tamaño que cambian de forma dinámica. De esta forma el tipo de contenido no es importante para la base de datos, solo la clave y el valor que tiene asociado. Son muy eficientes para lecturas y escrituras, además de que pueden escalar fácilmente particionado los valores de acuerdo a su clave; por ejemplo aquellos cuya clave está entre 1 y 1000 van a un SERVIDOR, los de 1001 a 2000 a otro, etc. Dentro de estas bases de datos podemos encontrar a BigTable de Google, SimpleDB de Amazon, Cassandra, Hadoop, Riak, Voldemort y MemcacheDB entre otras.
- Basada en Documentos: estas almacenan la información como un documento (generalmente con una estructura simple como JSON o XML) y con una clave única. Es similar a las bases de datos Key-value, pero con la diferencia que el valor es un fichero que puede ser entendido. Si el servidor entiende los datos, puede hacer operaciones con ellos. De hecho varias de las implementaciones de este tipo de bases de datos permiten consultas muy avanzadas sobre los datos, e incluso establecer relaciones entre ellos, aunque siguen sin permitir joins. Podemos encontrar a MongoDB y CouchDB entre las más importantes de este tipo.
- Orientadas a Grafos: Hay otras bases de datos que almacenan la información como grafos donde las relaciones entre los nodos son lo más importante. Son muy útiles para representar información de redes sociales. De hecho, las relaciones también pueden tener atributos y puedes hacer consultas directas a relaciones, en vez de a los nodos. Además, al estar almacenadas de esta forma, es mucho más eficiente navegar entre relaciones que en un modelo relacional. Obviamente, este tipo de bases de datos sólo son aprovechables si la información en cuestión se puede representar fácilmente como una red. Encontramos a Neo4J entre otras.
- Orientadas a Columnas: guardan los valores en columnas en lugar de filas. Con este cambio ganamos mucha velocidad en lecturas, ya que si se requiere consultar un número reducido de columnas, es muy rápido hacerlo pero no es eficiente para realizar escrituras. Por ello este tipo de soluciones es usado en aplicaciones con un índice bajo de escrituras pero muchas lecturas. Por ejemplo, Cassandra.
3.2. Neo4J
Neo4J es un software de código abierto que le permite utilizar base de datos orientada a grafos y está constituido la siguiente configuración:- Nodos: son las entidades que vamos a representar.
- Relaciones: representan las asociaciones entre entidades y siempre poseen un nodo de inicio y otro de destino.
- Propiedades: representan los atributos que se pueden almacenar tanto en los nodos como en las relaciones.
3.2.1 Neo4J en el detalle
El lenguaje utilizado por Neo4J está basado en Java y mantiene el grafo en disco en el interior de una estructura optimizada. El grafo en cuestión será:- Dirigido: cada arco posee una dirección.
- Completamente etiquetado: cada nodo tiene un nombre que lo identifica y cada arco posee una clase.
- Sujeto al atributo: es posible memorizar los pares nombre-valor sobre arcos y nodos.
- Recovery: recupera la información después de haberla perdido a causa de un error.
- 2-phase-commit: protocolo para aplicaciones distribuidas que permite mantener la propiedad ACID en las transacciones.
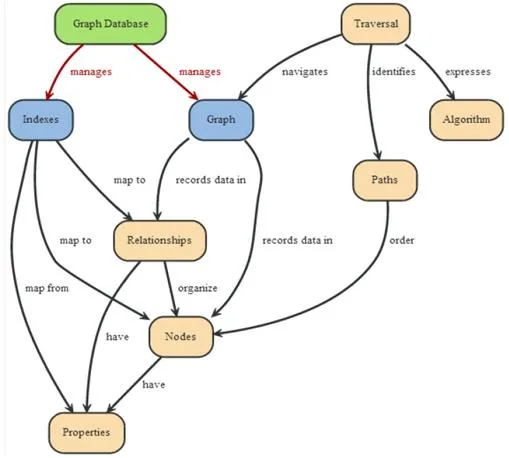
- Nodo: contiene una entidad de nuestro problema y debe estar asociado a un grafo. Dentro de él podremos definir tanto propiedades como pares clave-valor y quedarán contenidos en el interior del mismo nodo. Aparte de eso podremos utilizar estas propiedades para acceder al nodo que nos interese, es decir, podremos utilizar en las consultas para encontrar los nodos deseados. Además podemos acceder a los nodos a partir de dos métodos diferentes: índices y traverse que explicaré a continuación.
- Relación: representa la relación existente entre los nodos. Cada relación tiene un tipo que puede ser usado como método de búsqueda para cuando se realiza una búsqueda con índice. En cambio el método traverse no lo podríamos aplicar aquí porque sirve para consultar los nodos. Para obtener los nodos a través de los índice tenemos que indicar si queremos obtener el nodo de inicio o el final de la relación.
- Índice: se utiliza para obtener un conjunto de nodos o relaciones a través consultas a la base de datos con parámetros asociados al objeto o a los objetos que queramos obtener.
- Traverse: se utiliza para consultar a la base de datos de un nodo dado. Puede analizar las relaciones entrantes y saliente a través de un algoritmo (para coger solamente las relaciones interesadas) y definir incluso el número de saltos (hop) que el algoritmo tiene que cumplir como máximo.
3.2.2 Ejemplo gestión de base de datos Neo4J
Para entender mejor cómo funciona Neo4J, también hemos construido y explicado un programa sencillo que nos permite aprender todo lo que tenemos que saber antes de escribir algoritmos más complejos como las de las funciones de Ranking para el análisis de los genes.Ejemplo
Como nuestro algoritmo va a trabajar con grafos de genes, hemos considerado que el ejemplo ideal para mostrar el funcionamiento básico de Neo4J podría ser un simple algoritmo que trabaje con un árbol genealógico con el siguiente enunciado: Víctor ha adoptado una hija. El primer paso va a ser definir todas las variables que queremos utilizar durante nuestro algoritmo e inicializar la base de datos de Neo4J. Para ello creamos una variable HashMap <p ,String> con todas las propiedades que creamos oportunas para nuestra aplicación, que en mi caso considero que son:
HashMap properties ;
properties.put("online_backup_enabled","false");
properties.put("keep_logical_logs","false");
properties.put("db.mapped_memory","300M");
properties.put("relationshipstore.db.mapped_memory","300M");
properties.put("propertystore.db.mapped_memory","600M");
properties.put("propertystore.db.strings.mapped_memory","500M");
properties.put("propertystore.db.arrays.mapped_memory","0M");
Posteriormente tenemos que iniciar la base de datos. Para ello utilizaremos el método EmbeddedGraphDatabase() que necesita como primer parámetro la ruta donde queremos almacenar la información y como segundo parámetro opcional las propiedades que deseamos otorgarle a la base de datos. Inmediatamente después de crear el sistema, desde la documentación de Neo4J [neo4j.com 2014]recomienda utilizar el método registerShutdownHook que ha sido implementado para cerrar correctamente la base de datos cuando termine la máquina virtual de java. Entonces como resultado obtenemos:
graphDb = new EmbeddedGraphDatabase(DB_PATH,properties);
registerShutdownHook( graphDb );
El siguiente paso en Neo4J es definir las relaciones que vamos a utilizar durante todo nuestro programa indicando su nombre y que será un tipo de relación:
enum RelTypes implements RelationshipType { HIJA }
Con la relación creada, el siguiente paso es crear los nodos del padre y la hija, con información acerca de ellos, por ejemplo su nombre. Para ello simplemente tendremos que realizar los siguientes pasos: - Primero crear los nodos:
Node padre;
padre = graphDb.createNode();
Node hija;
hija = graphDb.createNode();
padre.setProperty( "nombre", "Victor " );
hija.setProperty( "nombre", "Andrea" );
Hecho esto, ahora nos queda establecer la relación definida como “HIJA” entre el nodo padre y el nodo hija:
relacion = padre.createRelationshipTo( hija, RelTypes.HIJA );
En Neo4J también se permite añadir propiedades a las relaciones como, por ejemplo, decir que la hija es adoptada:
relacion.setProperty( "tipo", "adoptada" );
Como resultado hemos obtenido la figura [3.4] , es decir, un grafo en el que tanto Víctor como Andrea los hemos representado con dos nodos y su relación familiar con un arco de nombre “HIJA” que los une. Para almacenar la información de que es adoptada, simplemente hemos añadido una propiedad al arco que indica esta característica. Otra opción que podríamos utilizar es crear más relaciones del tipo hija natural e hija adoptada que nos permitirá obtener el mismo resultado sin dar propiedades. En nuestro ejemplo vemos muy importante acceder de forma rápida a los nodos que identifiquen las hijas de Víctor y para ellos es más eficiente la primera opción ya que con una única consulta podemos encontrar a todas sus hijas. 
Relationship r;
padre.getSingleRelationship(RelTypes.HIJA,Direction.OUTGOING );
print("TIENE UNA HIJA DE NOMBRE:")print(r.getEndNode().getProperty("nombre"));
Otro punto muy interesante de Neo4J es el acceso de los nodos a través de los índices. Para ello nuestra base de datos nos permite definir una estructura que se llama Index. Una vez ha sido creada solamente le tenemos que indicar cuál va ser su finalidad, es decir si va a ser utilizado para acceder a nodos o relaciones y un nombre identificativo para acceder a ella.
public static Index <Node> padreindice;
padreindice=graphDb.index().forNodes("nombre");
Con esta estructura definida, tenemos que añadirle los nodos que queremos indexar y la llave para acceder a ellos, como por ejemplo:
padreindice.add(padre,"nombre","Victor ");
Una vez acabado este procedimiento, podremos utilizar la función get() que nos proporciona la interfaz Index para obtener el nodo que deseemos de forma rápida e eficiente.
Node recuperar=padreindice.get("nombre", "Victor ").getSingle();
El último tipo de consulta que nos queda por conocer es el método traverse. Como habíamos dicho, se utiliza para obtener los nodos que necesitemos de forma más rápida que con los índices. Para elaborar este tipo de consultas tenemos primero que aplicarle el algoritmo de búsqueda ( en nuestro ejemplo hemos aplicado breadth first que consiste en analizar los nodos que se encuentran más cerca al nodo de inicio), la profundidad de la búsqueda, el tipo de relación y que nodo nos interesa (el inicial o el final ). Como resultado nos proporciona una serie de vértices que tendremos que recorrer para obtener su información
Traverser t = recuperar.traverse(Order.BREADTH_FIRST,StopEvaluator.DEPTH_ONE,
ReturnableEvaluator.ALL_BUT_START_NODE,RelTypes.HIJA, Direction.OUTGOING);
Para acabar tenemos que explicar cómo usa las transacciones Neo4J. Podemos observar que la mayoría del código existen estas dos líneas de código.
Transaction tx = graphDb.beginTx();tx.success();tx.finish();
Su finalidad es permitir al usuario realizar operaciones sobre la base de datos sin ningún tipo de control. En el momento en el cual deseemos almacenar los cambios, llamaremos al método success() y comprobará que las modificaciones no rompe ninguna regla de integridad del sistema. 3.3. Titan cassandra
Titan Cassandra es un software de código abierto que le permite utilizar base de datos distribuidas orientada a grafos y está constituido la siguienteconfiguración:- Nodos: son las entidades que vamos a representar.
- Relaciones: representan las asociaciones entre entidades y siempre poseen un nodo de inicio y otro de destino.
3.3.1 Titan Cassandra en detalle.
Titan Cassandra mantiene el grafo en disco en el interior de una estructura orientada para ser distribuida. El grafo en cuestión será:- Dirigido o no dirigido: existen dos implementaciones posibles, construir un grafo donde cada arco posee una dirección o construir otro en que todos los arcos tengan las dos direcciones.
- Completamente etiquetado: cada nodo tiene un nombre que lo identifica y cada arco necesita un nombre para crearse.
- Sujeto al atributo: es posible memorizar pares nombre-valor sobre arcos y nodos.
- Escalabilidad horizontal. Titan Cassandra ha implementado diversos mecanismos para trabajar con grafos de gran tamaño a través de la capacidad de dividir el grafo analizado en proporción a la cantidad de clúster que posee nuestro sistema. Esta filosofía la ha aplicado también a las transacciones, permitiendo una cantidad simultánea de estas operaciones muy elevada e incluso permitiéndose dividirse en diferentes servidores.
- Consultas. Como en todos los gestores de bases de datos NoSQL, Titan Cassandra utiliza para realizar sus consultas y gestionar su estructura un lenguaje nativo, eficaz e intuitivo. Aparte de eso, el sistema puede realizar consultas por rangos de elementos, localización o búsquedas de llave- valor y así localizar tanto vértices como aristas.
- Soporta el Sistema Blueprints. Esta propiedad es muy interesante porque es un interfaz en código abierto que ofrece dos ventajas:
- Cualquier algoritmo escrito en Blueprints funciona de manera eficiente y sin ningún problema de adaptación en Titán. Esto también incluye a todo el software generado en código abierto de Tinkerpop, un grupo de trabajo que se encarga de desenvolupar nuevos sistemas para el manejo e interpretación de grafos.
- Cualquier consulta escrita para Titan se puede migrar a otros sistemas de base de datos orientado a grafos que acepta esta interfaz, así que no hay dependencia de un proveedor.
- Puede transformar un grafo en JSON. Esto es muy importante ya que hay muchas aplicaciones o incluso gestores de Bases de datos como MongoDB que aceptan este formato.
- Utiliza un método centrado a vértices. Ofrece la probabilidad al usuario de utilizar un método especial con el propósito de ordenar e indexar las aristas incidentes (y por tanto, vértices adyacentes) de un vértice de acuerdo con las etiquetas y las propiedades de las aristas. Utilizar bien este método cuando realicemos una consulta en sobre los vértices nos proporcionará una ventaja y las exploraciones lineales de aristas incidentes (O (n)) se pueden evitar y recorridos de gráficos más rápidos sobrevienen (O (1) u O (log n)).
- Sigue la filosofía NoSQL pero intenta optimizar el espacio utilizado. Proporciona una representación de disco optimizada para permitir el uso eficiente de almacenamiento y velocidad de acceso.
- Es código abierto bajo la licencia Apache 2.
- Nodo: Contiene una entidad de nuestro problema y debe estar asociado a un grafo. Dentro de él podremos definir pares clave-valor que quedarán contenidos en el interior del mismo nodo. Aparte de eso podremos utilizar estas propiedades para acceder al nodo que nos interese, es decir, podremos utilizar en las consultas para encontrar los nodos deseados. Cabe decir que Titan Cassandra otorga por defecto una id a cada nodo que se crea, siempre de valor numérico y puede ser utilizado para obtener los nodos que necesitemos.
- Relación: Representa la relación existente entre los nodos. Cada relación tiene un nombre que puede ser usado como método de búsqueda para cuando se realiza una búsqueda con índice. Aparte de eso Titan Cassandra también le otorga un id de forma automática.
- Índice: El sistema nos permite crear índices para acceder tanto a los vértices como a los arcos. Para ellos, existen dos métodos diferentes para diferenciarlos, uno se basa en etiquetas (labels) que permite almacenar índices a arcos que nos interesen. El otro, se basa en llaves (keys) que nos proporciona los diferentes métodos para acceder rápidamente a los vértices.
3.3.2 Ejemplo de gestión de base de datos Titan Cassandra
Una vez explicadas cada su estructura , el siguiente paso sería entender un poco como se puede construir un algoritmo en java basado en Titan Cassandra para entender todas las peculiaridades de su lenguaje nativo antes de escribir los algoritmos más complejos como los algoritmos para las funciones Ranking.Ejemplo
Como nuestro algoritmo va a trabajar con grafos de genes, hemos considerado el ejemplo utilizado en el sección 4.2.2 ya que es ideal para mostrar el funcionamiento básico de Titan Cassandra. Para recordar el enunciado vamos a implementar un simple algoritmo que trabaje con un árbol genealógico para representar el siguiente enunciado: Víctor ha adoptado una hija llamada Andrea. El primer paso de nuestra aplicación, va ser inicializar el sistema para poder almacenar el grafo. Para ello, primero aplicaremos el método erase( ) para asegurarnos que no exista otra base de datos con el mismo nombre que genere conflictos. Este método lo hemos implementado nosotros porque Titan Cassandra no posee ningún método para eliminar bases de datos. El algoritmo únicamente busca si existe la carpeta que utilizaremos para almacenar la base de datos y elimina todo su contenido. Si no utilizáramos este método, deberíamos realizar esta tarea manualmente cada vez que deseamos crear una nueva base de datos de prueba Una vez asegurado de que no existirá ningún conflicto, tendremos que configurar el sistema. Para ello, tendremos que indicarle sus características como por ejemplo, donde almacenará la información, si será un sistema local o distribuido o qué tipo de índices vamos a utilizar aplicando algoritmos como los siguientes:
BaseConfiguration config;
Configuration storage = config.subset(STORAGE_NAMESPACE);
configuring local backendstorage.setProperty(STORAGE_BACKEND_KEY,"local");
storage.setProperty(STORAGE_DIRECTORY_KEY, directory);
configuring elastic searchindexstorage.subset(INDEX_NAMESPACE).subset("search");
config.setProperty(INDEX_BACKEND_KEY, "lucene");
config.setProperty("local-mode",true);
config.setProperty("client-only", false);
config.setProperty(STORAGE_DIRECTORY_KEY, directory + File.separator + "es");
graph =TitanFactory.open(config);
Definida la estructura, nuestro grafo va a cumplir una interfaz llamada KeyIndexableGraph que posee métodos para crear índices y utilizarlo para consultas sobre vértices o arcos. Los métodos implementados para este fin son makeKey destinado para vértices y arcos y makeLabel solamente para arcos. Cuando aplicamos el método sortkey() a los índices, estamos aplicando la filosofía Vertex-Centric explicada anteriormente en el subapartado 3.2.1. Las consecuencias de esta acción sobre la base de datos es una redistribución de los arcos y vértices afectos de una forma especial y más eficientes para determinadas ocasiones. El único inconveniente es que para realizar esta tarea tenemos que invertir bastante tiempo.
.unique().make();tx.makeLabel("hija").manyToMany().sortKey().unidirected().make();Con toda esta información generada, el siguiente paso es crear los nodos del padre y la hija, con información acerca de ellos, por ejemplo su nombre. Para ello simplemente tendremos que realizar los siguientes pasos: - Primero crear los nodos:
Vertex padre = tx.addVertex(null);
Vertex hija = tx.addVertex(null);
padre.setProperty("nombre", "Victor");
hija.setProperty("nombre", "Andrea"); Hecho esto, ahora nos queda establecer la relación directa desde el nodo padre al nodo hija de la siguiente forma:
padre.addEdge("hija", andrea).setProperty("tipo", "Adoptada"); En Titan Cassandra también se permite añadir propiedades a las relaciones como por ejemplo decir que la hija es adoptada simplemente utilizando el método setProperty(). Como resultado hemos obtenido la figura [3.5] , es decir, un grafo en el que tanto Víctor como Andrea los hemos representado con dos nodos y su relación familiar con un arco de nombre “HIJA” que los une. Para almacenar la información de que es adoptada, simplemente hemos añadido una propiedad al arco que indica esta característica. Otra opción que podríamos utilizar es crear más relaciones del tipo hija natural e hija adoptada que nos permitirá obtener el mismo resultado sin dar propiedades. En nuestro ejemplo vemos muy importante acceder de forma rápida a los nodos que identifiquen las hijas de Víctor y para ellos es más eficiente la primera opción ya que con una única consulta podemos encontrar a todas sus hijas. 
Edge aux=victor.getEdges(Direction.BOTH,"hija");
Vertex resultado=aux.iterator().next().getVertex(Direction.IN);
print(resultado.getProperty("nombre")); Por último, como habíamos comentado, Titan Cassandra utiliza un sistema de transacciones que garantiza la integridad de los datos almacenados utilizando el siguiente código:
TitanTransaction tx = graph.newTransaction();tx.commit();
Por lo tanto, todas las sentencias que se ejecuten entre estas líneas solamente serán almacenadas en el caso de que no incumpla ninguna regla de integridad. 3.4. MongoDB
MongoDB es un software de código abierto que gestiona base de datos orientados a documentos. En un sistema normal de MongoDB siempre nos encontraremos estos tres elementos:- Documento: se define como los registros o conjunto de datos que gestiona el sistema. Cada uno de estas unidades está compuesto por pares “key-value” (clave-valor) donde la clave es el nombre del campo y el valor es su contenido, separados mediante el uso de “:” Como valor se pueden usar números, cadenas o datos binarios como imágenes o cualquier otra de par “key-value”.
- Coleccion: agrupación de documentos, las cuales se podría decir que son el equivalente a las tablas en una base de datos relacional. No posee ninguna limitación respecto a la cantidad
- Indice: El sistema de MongoDB está hecho para almacenar grandes cantidades de información y, por lo tanto, necesita de índices para localizar los documentos que necesitemos y buscar los valores deseados sin tener que recorrer todos los documentos.
3.4.1 MongoDB al detalle
MongoDB es un sistema de base de datos open-source y multiplataforma. Está escrito en C++, lo que le confiere cierta proximidad a los recursos de hardware de la máquina y le proporciona rapidez a la hora de ejecutar sus tareas. Además de tratarse de un software de licencia libre funciona en los sistemas operativos Windows, Linux, OS X y Solaris. Sus características más importantes a resaltar son:Almacenamiento orientado a documentos
Como hemos visto en la introducción MongoDB está orientado a la utilización de documentos del tipo JSON con un esquema dinámico llamado BSON, o Binary JSON, permite búsquedas rápidas de datos. Para hacernos una idea, BSON guarda de forma explícita las longitudes de los campos, los índices de los arrays, y demás información útil para el escaneo de datos. Es por esto que, en algunos casos, el mismo documento en BSON ocupa un poco más de espacio de lo que ocuparía de estar almacenado directamente en formato JSON. Este inconveniente siempre existe en los sistemas NoSQL, ya que realmente tiene una filosofía basada en el almacenamiento barato, y como consecuencia defiende que es mejor utilizar más memoria si así se introduce un considerable incremento en la velocidad de localización de información dentro de un documento. Un ejemplo de este documento es la figura [3.6]
Soporte Full index
Los índices en MongoDB poseen una estructura en árbol llamada B-Tree. Es decir, que los datos se guardan en un árbol cuyos nodos pueden tener un número múltiple de hijos, pero manteniendo todos ellos balanceados. Esto incrementa la velocidad a la hora de buscar y también a la hora de devolver resultados ya ordenados. De hecho MongoDB es capaz de recorrer los índices en ambos sentidos, por lo que con un solo índice, podemos conseguir ordenación tanto ascendente como descendente. Un detalle importante que nos puede ayudar a mejorar nuestra eficiencia en el rendimiento es que los índices los utilicemos con campos que posean gran cardinalidad. Cuando creamos un índice y vamos rellenando el árbol con la información que queremos indexar, va introduciendo los elementos en determinadas posiciones del árbol dependiendo del contenido que se encuentre en el campo. Por lo tanto cuanta más diversidad posea la información indexada, más eficiente será el índice. En otro sentido, el sistema nos ofrece diferentes tipos de índices. Primero, MongoDB indexa todos los documentos con la clave _id (explicada anteriormente), los que nos permite acceder rápidamente a ellos y evitar tener que recorrer toda la colección para encontrarlos. Después también nos ofrece índices geoespaciales, es decir, permite la indexación de la información basado en la localización. Por último también permite indexar campos de un documento, para acceder a ellos sin necesidad de recorrer todo el documento.Escalabilidad horizontal
En sistemas tradicionales RDBMS, para mejorar el rendimiento de la base de datos se adquiere una máquina más potente (escalado vertical) mientras que en MongoDB funciona mejor el escalado horizontal (incrementar número de máquinas)Consultas
Otros puntos fuertes a destacar de MongoDB son su velocidad y su rico pero sencillo sistema de consulta de los contenidos de la base de datos. Siguiendo la filosofía del NoSQL explica anteriormente, ha conseguido alcanzar un nivel de rendimiento y funcionalidad óptimos, incorporando tipos de consulta del sistema relacional, pero sin sacrificar el rendimiento.Map y Reduce
MongoDB nos permite utilizar funciones Map y Reduce para seleccionar los atributos que nos interesan de los datos, agregarlos, unificarlos y simplificarlos como deseemos. Esto es algo habitual en muchos sistemas NoSQL, y en algunos casos es incluso la única forma posible de consultar datos.Ausencia de transacciones
Realmente esta no se sabe si es una virtud o una deficiencia. Que MongoDB no tenga ningún sistema para comprobar que los datos introducidos corrompen la base de datos hace que los algoritmos implementados sean más rápidos pero el gestor de la base datos no bloqueara los “commits” con información errada.3.4.2 Gestión de la base de datos MongoDB
Una vez explicadas cada una de sus principales virtudes y deficiencias, el siguiente paso sería entender un poco como se puede construir un algoritmo en Java basado en MongoDB y así conocer su lenguaje nativo antes de escribir los algoritmos más complejos .Ejemplo
Como nuestro algoritmo va a trabajar con grafos de genes, hemos considerado el ejemplo utilizado en el apartado 4.4.2 ya que es ideal para mostrar el funcionamiento básico de MongoDB. Para recordar el enunciado vamos a implementar un simple algoritmo que trabaje con un árbol genealógico para representar el siguiente enunciado: Víctor ha adoptado una hija llamada Andrea. El primer paso va a ser definir todas las variables que queremos utilizar durante nuestro algoritmo e inicializar la base de datos de MongoDB. Para ello primero tendremos que saber dónde está situado el servidor que tiene almacenada la base de datos de MongoDB y conectar nuestro programa:
MongoClient conn = new MongoClient( "localhost" , 27017 );
Una vez conectado tenemos que crear o buscar la base de datos que vamos a utilizar. En el caso del ejemplo, primero compruebo que no hay nada en la base de datos y si hay lo borro y después se crea la estructura. Primero la base de datos con el nombre “Árbol” utilizando el método getDB().Este método busca en el servidor si existe alguna base de datos con ese nombre, si la encuentra la devuelve sino crea una nueva con ese nombre. Después creamos la colección Familia dentro de la base de datos “Árbol” que almacena todos los Documentos necesarios para su implementación. Para ello el método getCollection("Familia") aplicado a la variable db nos enlazara a la colección Familia si existe o la creara en caso contrario.
DB db;DBCollection coleccionfamilia;
for (String s : conn.getDatabaseNames()) {
conn.dropDatabase(s);
}
db = conn.getDB("Árbol"); //creamos la base de datos
coleccionfamilia=db.getCollection("Familia");
El último paso para acabar la construcción de nuestro sistema es crear un índice para acceder rápidamente a nuestro documento. Los índices en MongoDB se crean aplicando a la colección deseada el método createIndex() y pasándole como parámetro la llave para acceder al campo indexado que queremos junto con el valor 1 o -1 que indican si queremos ordenarlos de forma ascendente o descendente respectivamente.
this.coleccionfamilia.createIndex(new BasicDBObject("name", 1));
Una vez hemos creado toda la estructura que queremos utilizar el siguiente paso sería almacenar la información dentro de la colección. Para ello tenemos que recordarnos que MongoDB trabaja con Documentos en formato JSON. Para ello, Java pone a nuestra disposición unas librerías que trabajan este formato que nos permitirán crear y comprobar la estructura antes de introducirla en la base de datos. Con este fin, hemos implementado un objeto JSONObject llamado Víctor que dentro tiene almacenado como propiedades el nombre de la persona a quien representa, es decir, Víctor y las hijas que posee. Este último campo ha sido implementado como un JSONArray, es decir estará formado por un Array de LinkedHashMap donde cada uno de los elementos introducidos deberá contar con el nombre de las niñas y el tipo de relación que mantiene con Víctor (natural o adoptada por ejemplo).
JSONObject victor=new JSONObject();
JSONArray hijas=new
JSONArray();
this.victor.put("Nombre","Victor");
LinkedHashMap andrea=new LinkedHashMap();andrea.put("nombre","Andrea");
andrea.put("tipo", "adoptada");
this.victor.put("hijas",andrea);
familia.add(victor);
Como resultado de este trabajo hemos obtenido la figura [3.8] . que se muestra a continuación. 
Object classaux = com.MongoDB.util.JSON.parse(x.toString());
DBObject dbObjaux = (DBObject) classaux;
this.coleccionfamilia.insert(dbObjaux);
Una vez finalizado el proceso podemos observar el resultado real en la figura 8, es decir, un documento que representa a la persona Víctor y la cantidad de hijas que posee y la relación que los une. Lo importante de esta figura es que realmente en este documento hemos almacenado de forma fácil e intuitiva la misma información que en el grafo de la figura [3.9] . 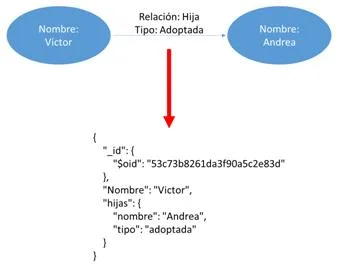
BasicDBObject query=new BasicDBObject("Nombre","Victor");
System.out.print("Victor TIENE UNA HIJA DE NOMBRE:");
DBCursor f =coleccionfamilia.find(query);while ( f.hasNext()) {
print((f.next().get("hijas")).get("nombre"));
} 3.5. Cassandra
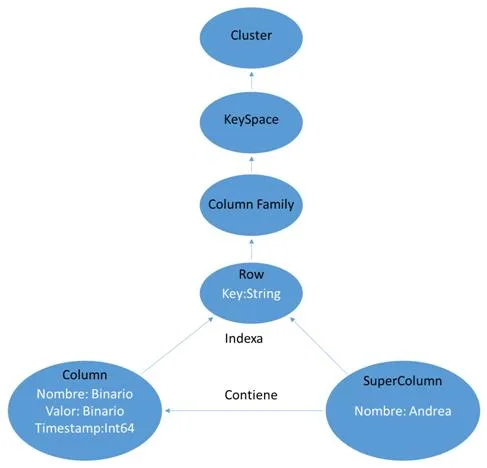
Cassandra es un software de código abierto que gestiona base de datos orientados a columnas. En un sistema normal de Cassandra siempre nos encontraremos estos elementos:- Column: Es el elemento más básico de la base de datos y se asemeja al concepto de campo en las bases de datos relacionales. Tiene una estructura muy parecida a un array asociativo ya que cada Column consta de los siguientes tres campos:
- Nombre: array de bytes que se utiliza para acceder al valor almacenado.
- Valor: array de bytes que puede contener cualquier tipo de información.
- Timestamp: variable int64 donde se almacena la última vez que fue modificada la Column. No hay restricciones con respecto a lo que pueden contener los campos. Podemos introducir tanto en el nombre como en el valor los campos que nosotros queramos, ya que como en todos los sistemas NoSQL no existe restricciones en la información almacenada.
- SuperColumn: Es una agregación de n-columns que puede referenciarse por un nombre. Es un recurso bastante útil, ya que permite tener una colección de valores asociados a un valor. De igual forma que antes, no hay restricción acerca de lo que los atributos pueden contener.
- Row : Es una agregación de columns o super columns que se referencian con un nombre contenido dentro de un String. Ese nombre es la clave (key) que identifica de forma unívoca a un registro. En este punto es importante no confundir un row con una super column. La row es sólo una palabra clave, no es una estructura, carece de atributos como las super column.
- Column Family: Es una agregación de rows referenciada por un String. Es una idea similar a una Tabla en el modelo RBDMS.
- KeySpace: Es una agregación de column families que puede referenciarse por un nombre. No es una estructura con atributos, es tan sólo otro contenedor al que se accede por un String. Si buscamos el equivalente con los modelos RBDMS sería una idea similar a los esquemas (conjunto de tablas) en el modelo relacional.
- Cluster: Es el elemento de más alto nivel que puede referenciarse por un nombre. Es de naturaleza más física que los anteriores, más relacionado con el hardware, ya que agrupa los nodos (máquinas) sobre los que se ejecuta Cassandra. Puede contener uno o más keyspaces.
3.5.1 Cassandra al detalle
Cassandra es una base de datos de código abierto, desarrollada en Java y multiplataforma. Su principal característica es la fusión de Dynamo, de Amazon con BigTable, de Google, siendo ambas implementaciones de código cerrado. Si buscamos un poco de historia sobre este sistema, nos daremos cuenta que fue desarrollado como respuesta a los problemas del Big Data descrito en la introducción. Facebook tenía una necesidad para mejorar el rendimiento del motor de búsquedas, concretamente las relacionadas en la comunicación entre usuarios (“Inbox Search“) ya que esta funcionalidad implica gestionar un gran volumen de datos, con una perspectiva de crecimiento muy alta (tenemos que pensar que el boom de las redes sociales se produjo después de la implementación de Cassandra). Para solucionar este problema se diseñó este sistema donde sus principales características fueran: consultas altamente escalables, horizontales y con un coste económico rentable. Posteriormente de invertir Cassandra, en 2008 la licencia de Cassandra fue liberada por Facebook, pasando a ser de código abierto, y actualmente es mantenido por Apache. Esta característica es la que hace de Cassandra una base de datos NoSQL realmente interesante, ya que combina lo mejor de Dynamo (consistencia eventual) con lo mejor de BigTable (familias de columnas) de forma gratuita. Las consecuencias de esta historia las podemos observar en las siguientes características que posee el sistema:- Tolerancia a los fallos: Los datos se replican automáticamente a varios nodos. Perder un nodo no causa la baja del clúster.
- Esquema flexible: Ya hemos comentado anteriormente que columns, super columns y column families puede almacenar los valores que deseemos.
- Simétrica: Todos los nodos en el clúster son idénticos lo que evita los problemas relacionados con los cuellos de botella.
- Escalable horizontalmente: Como ya habíamos comentado, es normal que los sistemas NoSQL aumente su eficiencia a medida que se introducen más servidores en el sistema
- Apoyo a grandes de datos: La capacidad para escalar a cientos de gigabytes de datos.
3.5.2 Gestión de la base de datos Cassandra
Una vez explicadas cada una de sus principales virtudes y deficiencias, el siguiente paso sería entender un poco como se puede construir un algoritmo en Java basado en Cassandra y así conocer su lenguaje nativo antes de escribir los algoritmos más complejos .Ejemplo
Como nuestro algoritmo va a trabajar con grafos de genes, hemos considerado el ejemplo utilizado en la sección 4.4.2 ya que es ideal para mostrar el funcionamiento básico. Para recordar el enunciado vamos a implementar un simple algoritmo que trabaje con un árbol genealógico para representar el siguiente enunciado: Victor ha adoptado una hija llamada Andrea. El primer paso va a ser definir todas las variables que queremos utilizar durante nuestro algoritmo e inicializar la base de datos de Cassandra. Para ello primero tendremos que saber dónde está situado el servidor y conectar nuestro programa mediante el método connect():
connect("127.0.0.1");session = cluster.connect();
Una vez realizado la conexión, la variable sesión nos permitirá realizar modificaciones o crear estructuras dentro del sistema. Podemos ver como en el ejemplo la hemos utilizado para realizar cualquier tarea como por ejemplo definir las estructuras de nuestra base de datos para: - Eliminar si existe un Keyspace llamado Familia y evitar posibles conflictos.
DROP KEYSPACE IF EXISTS Familia; - Crear, si no existe, un Keyspace llamado Familia. Cassandra permite otorgar características a nuestro Keyspace, para poder optimizar más la eficiencia del sistema. En nuestro caso hemos indicado que pertenece a la clase SimpleStrategy (recomendado para base de datos con un único servidor) y formado por un nodo.
CREATE KEYSPACE IF NOT EXISTS Familia WITH replication = { 'class':
'SimpleStrategy', 'replication_factor' : 1 };
CREATE TABLE Familia.perez (nombre text PRIMARY KEY, hijas map<text,text>);
INSERT INTO Familia.perez (nombre , hijas) VALUES (“Victor”, { "nombre": "Andrea","tipo": "adoptada"});
Como resultado hemos obtenido la [3.11] , es decir, una estructura en columnas que representa a la persona Víctor, la cantidad de hijas que posee y la relación que los une. Podemos observar se cumple una de las principales características de Cassandra ya que hemos almacenado de forma fácil, intuitiva y sin ninguna restricción la misma información que nos proporcionaba el grafo. 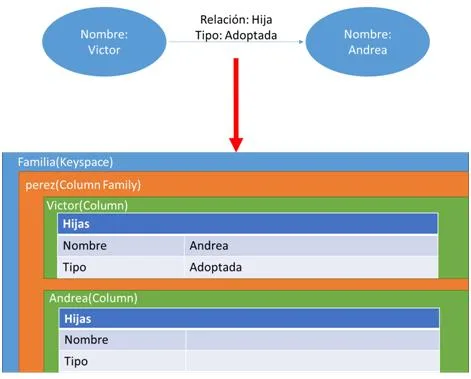
4. Funciones de predicción genética
Las funciones de ranking son aquellas funciones que procesan un conjunto de datos, predicen la probabilidad de que estén asociados entre sí sus elementos e indican el grado de pertenencia a una determinada característica, funciones o campo, es decir, una catalogación de los datos a partir de datos ya catalogados. Estas funciones son utilizadas en cualquier campo tecnológico o científico, ya que crean nuevos datos a partir de información previamente obtenida. En este proyecto se aplicarán las funciones de ranking al contexto de los genes, llamadas GFP (Gene Function Prediction).Las redes que se usan en los GFP siempre poseen una gran cantidad de datos y por lo tanto tendremos siempre problemas relacionados con la escalabilidad y el rendimiento. Si además unimos a esto que la cantidad de información crece día tras día, urge más estudiar y mejorar los procedimientos utilizados en los estudios. Frutos de estos acontecimientos, en pocos años la utilización de métodos computacionales existentes para identificar características y modelos han hecho catalogar millones de genes y crear redes de interacción genéticas que prometen mejorar los conocimientos en este campo. Para conseguir eso se utiliza un enfoque computacional que conduce a la definición de un gen y un conjunto de características que lo caracterizan. Después se aplican las técnicas de aprendizaje automático para identificar nueva información a partir de lo que sabemos [Pavlidis et al, 2001].La aparición de las redes de interacción de genes ha permitido analizar los datos en un contexto diferente que el de una sola dimensión. Una de estas redes es PPI (interacción proteína-proteína) y contiene asociaciones entre los genes humanos y los de otras especies [Aebersold et al, 2003]. Los datos que contiene se resumen en forma de grafos o documentos y en el apartado 4.1 se mostrará su estructura.El objetivo en los métodos de análisis es capaz de calcular la características asociadas con cada gen en los diferentes tipos de bases de datos enunciados en el apartado 2. Tenemos que analizar si es posible utilizar las bases de datos para esta finalidad y que método es más eficiente para estos sistemas.Como hemos comentado, existen varios enfoques para la resolución de estos problemas y empezaremos analizando si una red genética puede ser representada en grafos, documentos y columnas. Estas conclusiones las expondremos en la sección 4.1. La sección 4.2 hablaremos un poco sobre los métodos de lectura de datos sobre grafos. En la sección 4.3 analizaremos la función más interesante para nuestros sistemas, la función basada en kernels .En el última sección hablaremos del concepto de validación cruzada, utilizado función basada en kernels.4.1. Representación de los datos
Utilizaremos tres estructuras para la representar las redes de genes sus interrelaciones que serán:- Grafo para Neo4J y Titan Cassandra
- Documento para MongoDb
- Columnas para Cassandra
4.1.1 Grafo(Neo4J y Titan Cassandra)
Esta representación utilizara un grafo definido como el par G = (V, E) donde V representa los vértices, que corresponden a los genes, y E los arcos que los conectan, es decir, las relaciones entre los pares de genes. Este último puede estar asociado con un peso. La estructura en grafo se puede representar en una matriz de adyacencia que tiene los genes como una fila y columna de cabecera y el valor en que corresponde a la importancia relativa de la relación que los conecta. Esta matriz de adyacencia, que se identificarán con W, se transmite claramente es decir, el número de ceros que figuran en ella supera con creces el número de relaciones con diferentes pesos desde cero. Esto es debido al hecho de que cada gen se pone en correspondencia con todos los otros genes y por su naturaleza muchos genes no están conectados por relaciones, entonces, aparece un cero en la matriz. Por esta razón, tratamos de definir los algoritmos que hacen uso de esta característica. Un ejemplo de matriz W se encuentra en la figura [4.1] , en la que también se representa un ejemplo de formato denominada lista de adyacencia.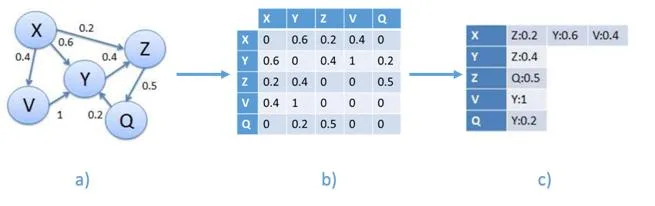
- [a)] ejemplo de un grafo que representa una red de genes. Cada nodo contiene un gen, cada arco contiene un peso que corresponde a la similitud entre genes.
- [b)] ejemplo de matriz W, correspondiente al grafo a)
- [c)] el formato de la matriz W en una lista de adyacencia ocupa menos espacio.
4.1.2 Documentos Bson (MongoDB)
La segunda opción para representar los datos es mediante Documentos. Más exactamente documentos del estilo JSON con un esquema dinámico llamado BSON, o Binary JSON, explicados ya en el apartado 2.4. Estos documentos estarán formados por campos clave-valor (o pongo key-value) que contendrá la información de cada vértice de nuestra red. Cada vértice red será representado con un documento que contendrá un campo llamado _id , otro con el nombre del gen y por último un array donde tendremos almacenado todos los nombres de los genes que está unido y su valor de similitud respecto a ellos.Como ejemplo, hemos realizado una conversión del grafo de la figura [4.2] a documento y hemos obtenido como resultado la figura 15 que es la siguiente:
4.1.3 Columnas (Cassandra)
La última de las tres representaciones es en columnas. Este formato es un poco especial ya que utiliza llaves (keys) para ir accediendo a la información. Para representar nuestra red en un contexto como este, tendremos que definir varios campos para situarlos. Primero un nombre al conjunto de todas las redes, por ejemplo como nuestros sistemas van a representar genes, podemos llamarlo “Genes” y representaría el Keyspace. El siguiente paso sería ir definiendo un poco más su contexto, como qué finalidad tendría nuestra red, ejemplo genesEspecieHumana y tendríamos definida la ColumnFamily. Y por último vendría cada una de las columns, correspondiente a los vértices de nuestra red. En esta última estructura es realmente donde está representado nuestro vértice porque por una parte tiene almacenado el campo clave-valor con el nombre del gen y por el otro un array formado por variables clave-valor con los vértices que posee una relación y el valor de similitud. En la figura [4.3] podemos observar esta estructura de forma gráfica para hacer entender mejor este formato.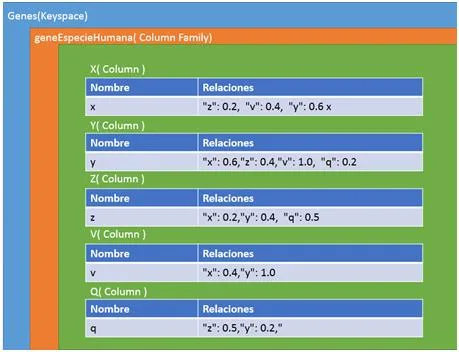
4.2. Los métodos utilizados en la lectura
A continuación se describen brevemente algunos de los principales métodos utilizados para predecir una función genética. Cada método sigue uno de estos enfoques:- Método directo: identifica las características de un gen a partir de las conexiones que hay en la red.
- Método asistido: identifica los genes similares a los analizados, así los genes analizados que carecen de catalogación heredan las propiedades de aquellos que son semejantes.
4.2.1 Método directo
Para entender los métodos directos vamos a utilizar la figura [4.4] mostrada a continuación.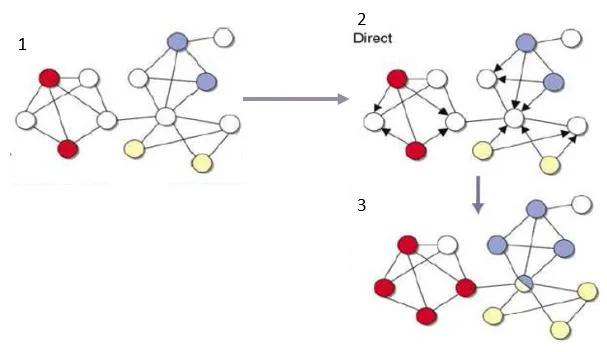
- [1] Se muestra una red en la que las funciones de determinados genes son conocidos (coloreados en colores diferentes) y otros (en blanco) que no son conocidos.
- [2] Se muestra los arcos con dirección para marcar cual es la influencia de un gen respecto a otro gen
- [3] Por último nos muestra en el diagrama 3 como los genes blancos cambian de color después de finalizar el proceso. También se puede observar en la imagen hay casos donde por ejemplo tenemos un gen puede tener asociado dos o más características.
- Neighborhood counting: este método [Schwikowski et al, 2000 ] predice funciones de los genes en base de las características ya conocidas de sus vecinos inmediatos. El problema que le hemos encontrado es su simplicidad y, aparte de eso, no tiene en cuenta la topología global de la red, solamente una parte muy pequeña.
- Grafo: Si pensamos que la mayoría de las redes son esencialmente un grafo es normal pensar que un método que se aproveche de esa característica nos será más beneficioso y, si encima, el método tiene en cuenta toda la topología de red (a diferencia del método anterior) conseguimos un algoritmo más eficiente. Buscando algoritmos en este contexto, hemos encontrado el método del grafo [Nabieva et al, 2005] donde se tiene en cuenta tanto la topología global, como el concepto de vecindad local para cada función genética que queramos analizar. El concepto básico es tratar a cada gen que ya tiene una función asociada como el nodo de origen de un diagrama de flujo. Después de la simulación, se asocia con cada gen carente de una función genética la probabilidad de que su función sea igual a la del gen de la salida. Este proceso se realiza para todos los genes que ya tienen una función asociada.
- Métodos probabilísticos: Se basan en el concepto de MRF (Markov Random Field) y se propusieron inicialmente en el artículo [Deng et al, 2003]. Con este método, la función asociada a un gen es semi-independiente de la de sus vecinos. Para saber si un gen tiene asociado una función genética tendremos que utilizar una fórmula logarítmica que tiene en cuenta el número de veces que aparece dicha función en la red y el número de genes asociados a la misma y que tienen relaciones con el gen de partida.
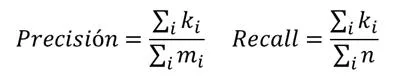
4.2.2 Método Asistido
Al igual que en la sección anterior, para entender los métodos asistidos voy a explicar a partir de la figura [4.6] mostrada a continuación.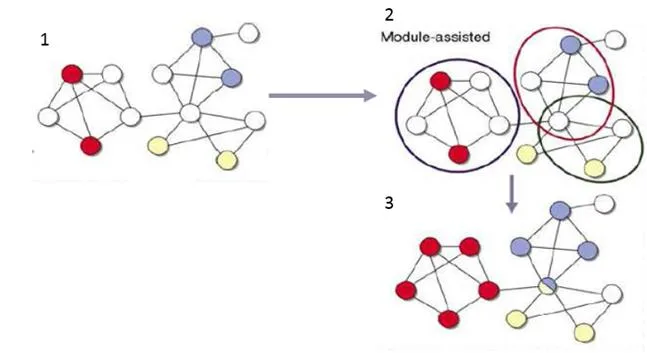
- [1] Se muestra una red en la que las funciones de determinados genes son conocidos (coloreados en colores diferentes) y otros (en blanco) que no son conocidos.
- [2] Se muestra una división de la red en conjuntos basados en la densidad.
- [3] Por ultimo nos muestra en el grafo 3 como los genes blancos cambian de color después de finalizar el proceso, cogiendo el color de aquel que predomina en el conjunto. También se puede observar en la imagen hay casos donde por ejemplo tenemos un gen puede tener asociado dos o más características.
Métodos genéricos
En estos métodos la elección del grupo se basa en la interacción entre los genes y la descomposición de la red en su topología. Uno de estos métodos es MCODE [ Bader et al, 2003 ] que se compone de dos etapas principales:- Se pesan los nodos de acuerdo con un coeficiente calculado a partir de la densidad de los nodos vecinos y, por lo tanto, tendrán un peso superior a los nodos con posean muchas conexiones.
- En el segundo paso se procesa el grafo dividiéndolo en secciones cuyos nodos posean entre ellos un gran número de conexiones
Método con clustering jerárquico
Este método utiliza un concepto de clustering jerárquico para repartir los datos en sus conjuntos. La ventaja de usar este enfoque se da por ser capaz de elegir la medida de similitud entre los pares de genes. Estos temas se tratan en [ Rives et al, 2003 ]Métodos de partición del grafo
Existen varios métodos que siguen este enfoque para grafos. En el artículo [ Spirin et al , 2003 ] por ejemplo, se exponen dos categorías bastante diferentes: los basados en SPC (Superparamagnetic clustering) [ Blatt et al , 1996 ] y basados en el método de Monte Carlo. En ambas categorías requieren como entrada el tamaño del cluster , pero la diferencia de los dos está en que el SPC funciona mejor en estructuras densamente conectadas, es decir, si llevamos esta información para los grafos, serían aquellos con una gran cantidad de arcos. Posteriormente, a partir de estos métodos se introdujeron otros nuevos algoritmos como RNSC (Restricted Neighborhood Search Clustering) [King et al, 2004] y el MCl[ Krogan et al, 2006 ]:- RNC es un algoritmo que dada una red divide sus elementos en diferentes conjuntos según una función de coste. Su procedimiento empieza con la creación de un clúster random y posteriormente procesa todos sus elementos y los reasigna basándose en la maximización de la función de coste. Para acabar el algoritmo filtra las diferentes agrupaciones según su tamaño y densidad.
- En otro sentido tenemos El MCL. Este algoritmo crea una matriz de adyacencia basado en la red de entrada e identifica las áreas altamente conectadas creando un conjunto con los elementos.)
Conclusión métodos asistidos
Entre los métodos asistidos que se han enumerado, MCL es el más robusto y con mejor rendimiento independientemente del grafo escogido posea datos reales o simulados. Las pruebas que se realizaron RNSC que era muy poco tolerante respecto a los parámetros de entrada. MCODE y SPC parecen inferiores si los comparamos con el algoritmo MCL, aunque tienen algunos aspectos a tener en cuenta como:- MCODE funciona mejor si los datos son aleatorios, proporciona una cantidad más baja de falsos positivos.
- SPC genera estructuras complejas para favorecer una mejor sensibilidad pero a costa de empeorar la especificidad.
4.3. Función de ranking basada en kernels
Entre todos los algoritmos analizados,el más eficiente para realizar nuestro estudio en grafos un método de ranking basado en kernels incluido en los métodos directos. Presenta siempre la siguiente fórmula: Su tarea consiste en asociar un número real a un gen que identifica el grado de pertenencia a una clase genética. Cuando este número es más alto, mayor es la probabilidad de que el gen pertenece a esa clase genética. El criterio de la distancia que utiliza una función de puntuación se refiere a un espacio de Hilbert, es decir, un concepto matemático definido como un espacio de producto interior que es completo con respecto a la norma vectorial definida por el producto interior [Wikipedia Hilbert 2014]. Si en el grafo no están presente arcos donde los nodos de salida y entrada sean idénticos (es decir todos los son iguales a 0 para cada ) se puede reducir la formula eliminando el primer término del algoritmo.Para facilitar el entendimiento de las operaciones, hemos expuesto un ejemplo en la figura [4.7] .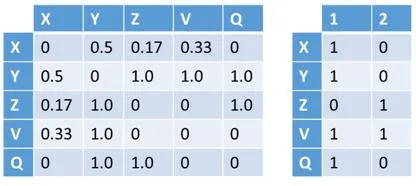
4.4. Técnicas de validación cruzada
Las técnicas de validación cruzada son utilizadas para evaluar el rendimiento de un algoritmo basado en el aprendizaje automático.Por lo tanto, podremos utilizar esta técnica para evaluar la eficiencia de las funciones de ranking basada en kernels. El aprendizaje automático se ocupa de los algoritmos basados en la observación de los datos existentes para la síntesis de nuevos conocimientos. El aprendizaje lo podremos obtener mediante información proporcionada por ejemplos, estructuras de datos o sensores, con la finalidad de analizar y evaluar las relaciones entre las variables observadas. La totalidad de los datos disponibles se llama dataset y se divide en dos partes: un training set y un test set . El training set representa los datos conocidos y se compone de un conjunto de elementos ya catalogados, mientras que el test set es un conjunto de elementos carentes de la catalogación. El algoritmo debe utilizar el conocimiento del conjunto training set para clasificar con él los elementos del test set. En algunos casos, sin embargo, podemos tener un conjunto training set con pocos elementos o el análisis llevado a cabo en el conjunto de training set ha durado demasiado tiempo. Estas situaciones se conoce como overfitting y aunque los elementos forman parte de la training set son correctos, pero la obtención de nueva información es difícil. Para evitar este inconveniente se han desarrollado técnicas de validación cruzada aplicadas en los programas expuestos en el capítulo 6. Las técnicas de validación cruzada requieren un conjunto de datos de tamaño considerable y proporcionan un criterio de pertenencia, de esta forma cada elemento se puede dividir en dos conjuntos diferentes, uno que tiene todos los elementos positivos y otro con los negativos. Si hablamos de los genes, el criterio de pertenencia podría ser por ejemplo la asociación del gen a una clase genética. En este punto, todo el dataset viene dividido en N conjuntos de tamaños similares llamados fold. El algoritmo se realiza en ciclos de N veces cambiando el fold puesto de ejemplo. Cada fold utilizado en un ciclo ser considerado como el test set y tiene el conjunto de los positivos y negativos. Una vez que este procedimiento ha terminado es el momento de realizar una estimación de todo el dataset y podremos realizar las estimación necesarias.Un ejemplo de esta técnica es la figura [4.8] , donde observar cómo hemos dividido los genes en dos conjuntos los positivos y los negativos a una determinada clase genética. Posteriormente, todos los genes se han dividido equitativamente en 5 folds.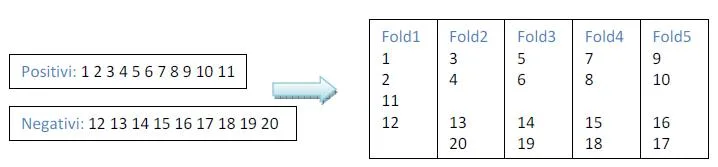
5.Herramientas y tecnologías usadas en el proyecto
En este apartado vamos a comentar las tecnologías empleadas para llevar a cabo el proyecto. Empezaremos en la primera sección eligiendo el lenguaje de programación que utilizaremos. En la segunda sección vamos a comentar que herramientas hemos empleado para elaborar el proyecto.5.1. Elección del lenguaje de programación
Tener que realizar la programación desde desde cero nos da la ventaja de poder elegir el lenguaje de programación que se va a utilizar. En este caso se plantearon inicialmente 3 lenguajes:- C++
- Python
- Java
5.2. Herramientas de desarrollo
Para desarrollar el proyecto se han utilizado las siguientes herramientas:- Eclipse: utilizado como entorno de programación en Java.
- Dropbox: como herramienta para guardar copias de seguridad del proyecto y a la vez tener un control de versiones de cada nueva funcionalidad.
- TexMaker: utilizado para realizar la memoria del proyecto.
5.2.1 Eclipse
Eclipse es un IDE muy útil en cuanto a la programación en Java se refiere, ya que proporciona muchísimas funcionalidades para que la programación sea mucho más cómoda. A continuación se nombran las características que han sido de más utilidad y que sin duda han reducido el tiempo empleado en el proyecto:- Editor de texto con resaltado de sintaxis.
- Compilación en tiempo real.
- Autocompletado inteligente.
- Plantillas para crear trozos de código, como nuevas clases, constructores, bucles, etc.
- Señalización automática de errores y avisos de compilación y sugerencias para resolverlos.
- Navegación entre clases y métodos.
- Generación automática de javadoc, jars y tgzs.
5.2.2 Dropbox
Dropbox es un servicio de alojamiento de archivos multiplataforma en la nube, operado por la compañía Dropbox. El servicio permite a los usuarios almacenar y sincronizar archivos en línea y entre computadoras y compartir archivos y carpetas con otros. Existen versiones gratuitas y de pago, cada una de las cuales con opciones variadas5.2.3 Texmaker
Texmaker es un editor gratuito distribuido bajo la licencia GPL para escribir documentos de texto, multiplataforma, que integra muchas herramientas necesarias para desarrollar documentos con LaTeX, en una sola aplicación Texmaker incluye soporte Unicode, corrección ortográfica, autocompletado, plegado de código y un visor incorporado en pdf con soporte de synctex y el modo de visualización continua.6. Desarrollo del Software
Como ya se ha comentado en el capítulo 1, el objetivo de este proyecto es implementar la función ranking en diferentes sistemas de gestión de bases de datos para comparar su eficiencia y rapidez en el manejo de grandes cantidades de información utilizando un ordenador sin grandes recursos hardware. En la etapa de diseño se intentará crear un diseño flexible y reutilizable de tal forma que permita extender y modificar la aplicación fácilmente. Esta parte se considera fundamental, ya que es probable que en el futuro se disponga de más operaciones para agregar a la aplicación que surja de este proyecto. De esta forma hemos dividido este apartado en varios subapartados como serán: 6.1 Donde analizaremos el análisis y el diseño del proyecto, 6.2 Implementación de la función score basada en kernels para cada una de las tecnologías expuestas, 6.2 Resultados, 6.3 Conclusiones6.1. Análisis y diseño de la función Score basada en kernels
Durante el inicio de esta etapa se ha prestado mucha atención a la distribución de los requisitos del proyecto en diferentes funcionalidades evitando complejidades innecesarias y crear clases o procedimientos inútiles, siempre intentando crear sistemas más fáciles de entender.6.1.1 Diagrama de casos de uso
Se ha comenzado definiendo los posibles casos de uso que puede realizar el usuario. De esta forma se ha obtenido la información sobre la funcionalidad que requiere el sistema. En la figura [6.1] se muestra el diagrama de casos de uso.
6.2. Implementación de la función Score basada en kernels
En este apartado del proyecto vamos a definir la implementación de la función Average Score. Para ello vamos a presentar los diferentes documentos que dispondremos como entrada para generar nuestras bases de datos en la sección 6.2.1 y el resultado final proporcionado en la sección 6.2.2. También expondremos en la sección 6.2.3 cómo hemos implementado los cálculos expuestos en el apartado 3 para obtener las probabilidades y cada uno de los detalles que hemos tenido que enfrentarnos en cada uno de los diferentes sistemas que hemos utilizado: Neo4J, MongoDB, Cassandra , Titan Cassandra.6.2.1 Formato de datos de entrada
Para generar la base de datos donde aplicaremos los cálculos utilizaremos los tres archivos de texto expuestos a continuaciónFile gene.txt
Contiene una columna con todos los genes con todos los nombres de los genes que vamos introducir en la base de datos. Podemos ver un ejemplo de fichero en la siguiente figura [6.2]
6.2.1.2 File geneclasse.txt
Contiene la matriz de adyacencia entre los genes y sus respectivas clases. Cuando en la matriz aparece un valor de 1 corresponde a que el gen de la fila pertenece a la clase de la columna y cuando aparece 0 viceversa. Esta matriz siempre cumplirá la siguiente característica: una clase es definida para un gen y este último puede pertenecer a varias clases. Como se muestra a continuación, en el archivo la primera fila contiene una lista con los identificadores para las clases. Después, las siguientes filas (separadas con un salto de línea) empiezan por el identificador del gen, y una listas de valores con 0 o 1 que nos informan si pertenece (1) o no pertenece (0) un gen a esa clase determinada. Podemos ver un ejemplo de fichero en la siguiente figura [6.3]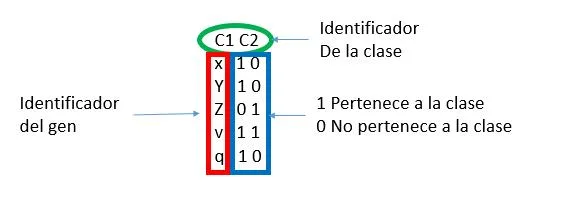
File gene-classe.txt
Contiene el valor de similitud entre un par de genes. Las dos primeras columnas obtendremos el identificador de los genes y en la tercera columna su valor de similitud, que siempre será mayor de 0 y menor o igual a 1. En la figura [6.4] se muestra un fichero de ejemplo: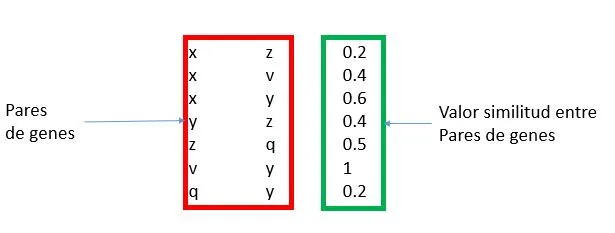
6.2.2 Explicación general del Algoritmo
La finalidad del código que vamos a implementar es generar bases de datos que representen redes de genes en 4 sistemas diferentes, utilizando para ello los tres archivos de entrada que se describen en la introducción de este capítulo. Con este fin los diferentes sistemas nos proporcionan una serie de posibles configuraciones, para optimizar al máximo los recursos de la máquina que disponemos y mejorar la eficiencia en servidores locales sin grandes recursos. Una vez clara estas configuraciones en cada situación, todos nuestros algoritmos siempre empezaran creando bases de datos vacías listas para introducir información y realizar todas las consultas necesarias. El segundo objetivo que tendremos que implementa, son los métodos que se encargará de introducir los vértices que representan los genes, las clases y los arcos que los relacionan entre sí. Para hacer esta tarea, podemos aplicar diferentes filosofías para aumentar más la robustez, la eficiencia o rapidez dependiendo de las necesidades del proyecto. En nuestro caso, nosotros hemos optado por utilizar una versión más robusta ya que si analizamos todo el algoritmo podemos comprobar que los dos últimos métodos requieren la búsqueda de vértices que ya han sido instanciados en los métodos anteriores, es decir, que si analizamos un poco más el código, sería más interesante realizar solamente la llamada de uno o dos métodos y reducir el número de búsquedas en la base de datos y, por lo tanto, disminuir los tiempos necesarios para crear la base de datos. Pero, realmente es preferible realizarlo de este modo porque al estar tratando con bases de datos tan grandes y con tantos elementos existe una probabilidad muy alta de que se produzca un problema en mitad de la ejecución, un fallo en la implementación o un error en la transición, y, en todos esos casos nos resultaría mucho más fácil solucionar el problema si resulta fácil entender la base de datos y en qué estado se encuentra. Los principales pasos para crear la base de datos en cada uno de los sistemas tienen muchas partes comunes que vamos a explicar en los siguientes subapartadosCreación de los genes
Este método se encarga de leer el fichero gene.txt (analizado en el apartado 6.2.1) para poder obtener la lista de genes con la que vamos a interactuar durante el programa. Nuestro algoritmo leerá el fichero de texto línea por línea para ir obteniendo y almacenado cada uno de los nombres de los genes que vamos a utilizar.Creación de las classes
Este método empieza leyendo la primera fila del archivo geneclasse.txt y aplica un split en ella, utilizando como elemento separador el espacio. El contenido obtenido corresponderá a todas las clases de genes posibles que posee nuestra base de datos y será almacenada esta información en un vector. El siguiente paso es recorrer el vector y con cada uno de sus elementos almacenados crear las estructuras que representen las clases.Creación de las conexiones entre genes y clases
Este método empieza igual que el anterior, leemos la primera fila del archivo geneclasse.txt y aplica un split en ella. El contenido será almacenado en un vector que después será utilizado en otra parte del algoritmo. Posteriormente, el método continuará leyendo el resto de información del documento, donde en cada una de las filas realizará el mismo procedimiento:- Realiza un split en la fila para obtener toda la información separada y accesible.
- Lee el primer elemento de la fila (el nombre de los genes) para poder realizar una búsqueda del gen que corresponde.
- Lee los siguientes elementos de la fila que se corresponden con la información de la tabla explicada en el apartado anterior y, por lo tanto, si la celda correspondiente tiene un valor 1, insertamos en la base de datos una relación que indique esta pertenencia del gen a la clase, y si el valor es 0, lo contrario.
- Realizaremos este proceso hasta acabar de leer todo el documento.
Creación de las conexiones entre genes
Este método se encarga de leer el fichero genegene.txt para obtener el valor de similitud entre dos genes, es decir, identificar genes en la base de datos con similares características. El archivo en cuestión está formado por una serie de filas donde, como hemos dicho en el apartado anterior, y mediante un split podremos obtener la información que nos interese y almacenarla en un vector. Los dos primeras partes tendrán almacenado los nombres de los genes que podremos utilizar posteriormente para realizar consultas en la base de datos. Y en la tercera parte del vector tendremos almacenado el valor en forma de entero que representa la similitud entre ellos. Este algoritmo tiene una característica especial. Durante la implementación surgieron problemas de fallos en la creación al tratar con cantidades muy grande de genes y por lo tanto cada vez que se alcanza las 10.000 relaciones analizadas se almacena la información nueva dentro de la base de datos y en caso de parada es necesario que realizar todas las transacciones desde el principio.6.2.3 Validación cruzada
El primer problema que hay que abordar antes de analizar la función de Score para una determinada clase es subdividir los genes en 5 folds. Para realizar los cálculos de una forma más sencilla y optimizada hemos decidido dividir cada uno de los folds en dos subestructuras que almacenen de forma separada los genes positivos y negativos. Todas estas estructuras las hemos creado e implementado en una clase especial llamada Fold donde su constructor será el encargado de realizar todo el proceso. Este método recibe como parámetro una referencia a una clase genética e implementará el pseudocódigo expuesto en la figura [6.5]
- n_fold : cantidad de folds que vamos a utilizar en nuestro algoritmo. Normalmente utilizaremos un valor 5 que es el valor estándar utilizado en la bibliografía consultada.
- positiveGenFold e negativeGenFold : Contendrán almacenados tantos ArrayList como número de fold utilicemos en nuestro algoritmo y su finalidad será ser rellenados con los genes positivos o los negativos respecto a una clase analizada.
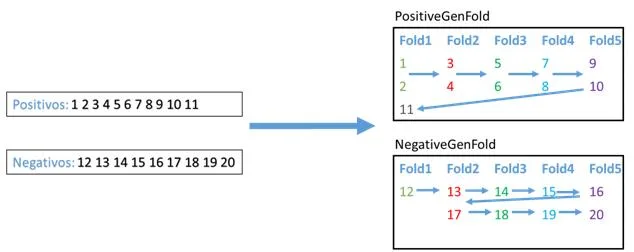
6.2.4 Función Score
Una vez que hemos terminado de crear los fold tenemos que proceder con el cálculo de la función de Score, representado en pseudocódigo en la siguiente figura [6.7]
- Obtener todos los genes que pertenecen a la clase investigada, excepto aquellos que ya están dentro del fold analizado. Podemos entender más fácil este proceso en la siguiente figura [6.8]
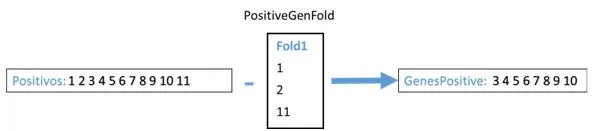
6.8 Representación de todos los genes que pertenecen a la clase investigada, excepto aquellos que ya están dentro del fold analizado - Recorremos el fold con el objetivo de aplicar el método average score a cada gen que hay almacenado. Como parámetros, este algoritmo necesita el gen que vamos a estudiar junto con la estructura GenesPositive para la clase a la cual puede pertenecer. Para mejorar el rendimiento del programa, hemos implementado el array de GenesPositivo como un hashset ya que las búsquedas de elementos en esta estructura tiene un coste constante y resulta mucho más beneficioso que un array. Respecto al método AvgScore cabe comentar que se encarga de simular los cálculos matemáticos expuestos en la sección 4.2. es decir simulara la función de ranking basada en kernels. El procedimiento empieza obteniendo el valor correspondiente al del gen analizado, es decir, el resultado de dividir 1 entre la raíz cuadrada del valor total de la suma de la similitud que tiene con todos los genes conectados. El siguiente paso es, para todos aquellos genes con los cuales está relacionado y pertenezcan a la clase analizada (está contenido en él has dado por parámetro), realizar el siguiente producto entre:
- el valor de del gen analizado, es decir,
- el valor del gen que está relacionado (se obtiene de la misma forma que el pero lo haces respecto al gen j, que sería en este caso aquel con el que se encuentra conectado).
- el peso de la relación que existe entre ellos.
- Un hastable rellenado con los pares de gen y probabilidad de pertenecer a las clases
- una lista con las clases que hay en la base de dato
- el nombre final que queremos otorgar al fichero.
6.3. Implementación en Neo4J
La primera parte del proyecto se basa en la implementación del algoritmo para Neo4J. Para realizar este experimento hemos utilizado la versión 1.8.2 obtenida del sitio oficial de Neo4J. Esta versión aun no permite la distribución de la información en diferentes servidores pero el equipo desarrollador trabaja en una futura versión que permita esta mejora sustancial.En el siguiente apartado explicaremos cómo hemos utilizado los diferentes mecanismos que nos proporciona Neo4J para resolver los algoritmos expuestos anteriormente. Los subapartados que nos vamos a encontrar son los siguientes:- Diagrama de clases
- Representación de la estructura de la base de datos
- Utilización del método validación cruzada.
- Implementación de la función Score.
- Diagrama de actividades.
6.3.1 Diagrama de Clases
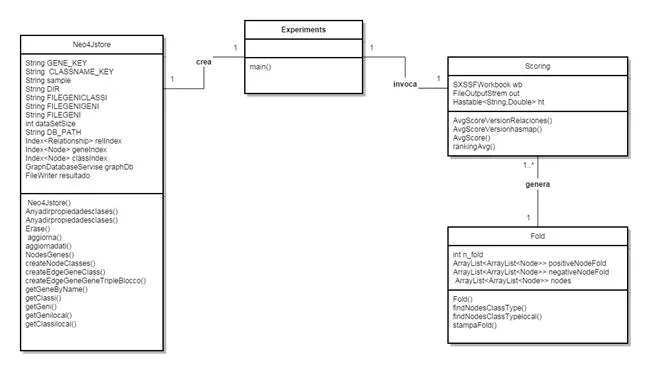
6.3.2 Creación de la estructura
Para crear una base de datos de Neo4J tenemos que diseñar su configuración a partir de la máquina donde realizamos el trabajo. Siguiendo la filosofía NoSQL hemos hemos optado por ejemplo evitar saturar el sistema ocupando mucha memoria, y evitando crear un backup o logs de estados que ocupen mucha memoria. Con la construcción finalizada, tendremos que implementar los métodos que se encargará de introducir los nodos que representan los genes, las clases y los arcos que los relacionan entre sí. Para hacer esta tarea hemos implementado los algoritmos expuestos en el apartado 6.2.2 que utilizara diferentes implementaciones para ver que opción proporciona una respuesta más rápida y eficiente. Como vimos en apartados anteriores, Neo4J es un sistema que gestiona nodos y relaciones. Cada nodo va a representar un gen de nuestra red, cuyo nombre figura en la propiedad “name” dentro del nodo. Cada vez que Neo4J crea un nuevo nodo le asocia una id automáticamente que comienza con 1 y se incrementa para cada nodo añadido. Esta id se convierte en índice automáticamente pero no ocurre lo mismo con la propiedad nombre. Si queremos aumentar la eficiencia en la búsqueda y obtención de nodos la forma más correcta de hacerlo es creando un índice (llamado gene) respecto al nombre, ya que utilizando este parámetro podremos crear consultas a la base de datos mucho más eficientes respecto al tiempo. Cabe señalar que tanto los nombres como los id que crea el Neo4J son únicos y por lo tanto a cualquier consulta que realicemos utilizando este parámetro obtendremos como respuesta un único nodo. Respecto a los nodos que van a representar las clases va a tener una estructura muy similar, la única diferencia con respecto a la creación de los nodos anteriores es que ahora el nombre de la clase se utilizará para crear el índice llamado “class”. Por último, las relaciones van a representar las conexiones que existen entre los genes y las clases de la red. En el caso que la conexión se produzca entre genes lo vamos a representar creando una relación “SIM” y cuando se produzca entre genes y clases la etiquetamos como “class”. Aparte de eso, en las relaciones de tipo “sim” almacenaremos el valor de la similitud entre los dos genes, creando una propiedad llamada “SIM”. Otros aspectos a comentar son las mejoras de rendimiento y robustez que hemos introducido en la implementación de este apartado. Para buscar los nodos que necesitamos en una relación utilizaremos los índices que hemos implementado ya que proporciona una disminución en los tiempos de búsqueda. Respecto a la robustez hemos introducido en todos los métodos que hemos trabajado las transacciones. Las transacciones es una herramienta que nos proporciona Neo4J para que solamente se guarden las modificaciones en el caso en que no se produzca o exista ningún error. El resultado de la implementación de este apartado se muestra en la siguiente figura [6.10]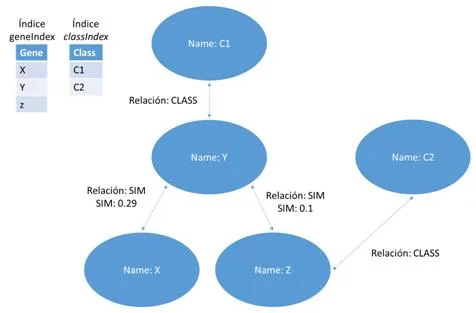
- : contendrá el valor explicado en la sección 6.2.2.
- type: indicará que el Vertex es un gen. Esta propiedad la utilizaremos en la implementación sin índices para buscar en la base de datos todos los genes que hay almacenados.
- Las relaciones entre los genes las almacenamos de dos formas diferentes para ver el rendimiento del sistema utilizando variables HashMap o ArrayList. Por ello crearemos:
- Relacionesmap: contendrá una variable Hashmap con las relaciones que posee en el nodo. Cada elemento tendrá almacenado como clave el nodo con el que existe la relación y el peso de la relación.
- Relaciones: Propiedad que almacena un ArrayList con el nombre de todas las relaciones.
- Relpesos: Propiedad que almacena un ArrayList con el peso de todas las relaciones.
- type: indicará que el Class es un gene. Esta propiedad la utilizaremos en la implementación sin índices para buscar en la base de datos todos los genes que hay almacenados.
- Positive: tendrá almacenado todos los genes que pertenece a este Nodo.
- Negative: tendrá almacenado todos los genes que no pertenecen a este Nodo.
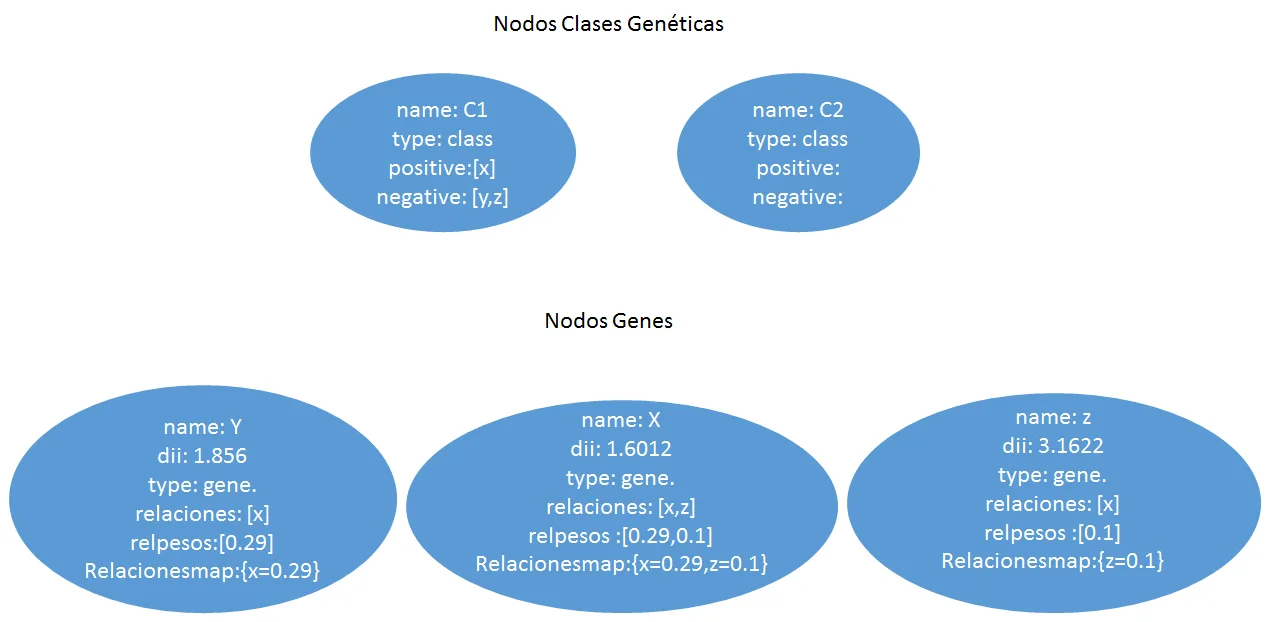
- Anyadirpropiedadesclases(): Se encarga de añadir a los nodos que representan las clases genéticas las propiedades type, positive, negative.
- Anyadirpropiedadesclases(): Se encarga de añadir a los nodos que representan los genes las propiedades type, relaciones, relpesos, relacionesmap.
- Neo4Jstore(String, int , int, String): Constructor de la clase. El primer parámetro que recibimos puede contener la siguiente información
- “crea”: Construye una nueva base de datos.
- “aggiorna”: Actualiza la base de datos
- “aggiornadati”: Actualiza datos que están dentro de la base de datos
- aggiorna(): Método para actualizar la base de datos.
- aggiornadati():Actualiza datos que están dentro de la base de datos
6.3.3 Validación cruzada
Las modificaciones que permiten adaptar este algoritmo (explicado en la sección 6.2) a las bases de datos de Neo4J son pocos, como por ejemplo que los arrays positiveGenFold e negativeGenFold van a gestionar Nodos. La única diferencia sustancial la vamos a encontrar en el método findNodesClassType() ya que es posible desarrollarla con diferentes filosofías. Los dos métodos de la clase Fold resultantes han sido:- findNodesClassType():Esta función se encargará de realizar rápidamente una consulta en la base de datos para buscar todos los arcos del tipo “class” que salgan del nodo proporcionado como argumento. Como podemos observar Neo4J es muy intuitivo, al ser una base de datos NOSQL orientada a grafos, nuestra consulta simplemente tendrá qué buscar todos los arcos salientes del tipo “class” que salgan del nodo de la clase analizada. Para realizar la consulta podremos utilizar parámetros como por ejemplo que el tipo de búsqueda utilizaremos en el grafo, que relaciones vamos a buscar, a qué profundidad o cuántos nodos vamos a analizar. El resultado será devuelto mediante una variable del tipo iterable y por lo tanto con un simple for podemos recorrerla y almacenar en un array el contenido de la búsqueda, es decir todos los genes positivos respecto la clase. Con esa información podremos obtener la lista de los genes negativos y, por lo tanto, todos los genes que no estén en esta lista, serán negativos respecto a la clase y tendremos también el array con los vértices negativos.
- findNodesClassTypelocal():Esta función utiliza la implementación más local de la información. Como no existen las relaciones, para obtener los genes positivos a la clase solamente tenemos que buscar la propiedad positive que contiene esta información. Una vez obtenida es almacenada en una variable y devuelta para realizar los cálculos necesarios. El mayor problema que nos vamos a encontrar para crear esta función es que Neo4J solamente puede gestionar String en las propiedades. La solución por la que hemos optado es utilizar las funciones replace() y split() de la librería String de Java para obtener los nombres de los genes y mediante la función getGeneByName obtener el gen deseado.
6.3.4 Score
En la clase Scoring las modificaciones también están relacionadas con el sistema de consultas de Neo4J.Primero de todo hemos implementado 3 métodos especiales para conseguir el correcto funcionamiento de la función Score que son:- setD(): este método recorre todos los genes de la base de datos para calcular el valor de que posee el gen analizado. Una vez que ha sido calculado lo almacena en el Nodo como . Para realizar esta tarea simplemente recorre todas las relaciones tipo “Sim” que posee el nodo analizado con el método getRelationships(). Este método de Neo4J va asociado a los vértices, recibe como parámetro el tipo de relación que estamos buscando y nos ofrece como resultado un iterador que recorre todas aquellas que están conectadas a nuestro nodo. El siguiente paso es ir cogiendo el valor de similitud almacenado anteriormente y sumar todos los valores para obtener el .
- AvgScore(): Con la función getRelationships() podremos recorrer todas las conexiones del gen analizado para obtener los valores de similitud y de los vertex que necesitamos.
- AvgScoreVersiónLocal() Este método utiliza las propiedades que hemos almacenado en el nodo y que contiene toda la información de sus relaciones. Podemos obtener el nombre de las conexiones a través de la propiedad “relaciones”, el valor de la propiedad llamada igual, el valor de similitud de sus conexiones a través de la propiedad “relpesos” y el valor Djj del Nodo con el método getGeneByName() dando como parametro el gen que nos interesa.
- AvgScoremap(): Misma metodología que el anterior pero ahora la información de las relaciones y el valor de la similitud la obtendremos de la misma estructura, un hashmap almacenado en la propiedad relacionesmap.
- rankingAvg() Función encargada de realizar los procedimientos para llevar los cálculos de ranking
6.3.5 Modelo de ejecución/navegación
Una vez descritos los posibles casos de uso que puede ejecutar el usuario junto con sus flujos de eventos vamos a describir la navegación que se puede realizar en la aplicación.figura [6.12]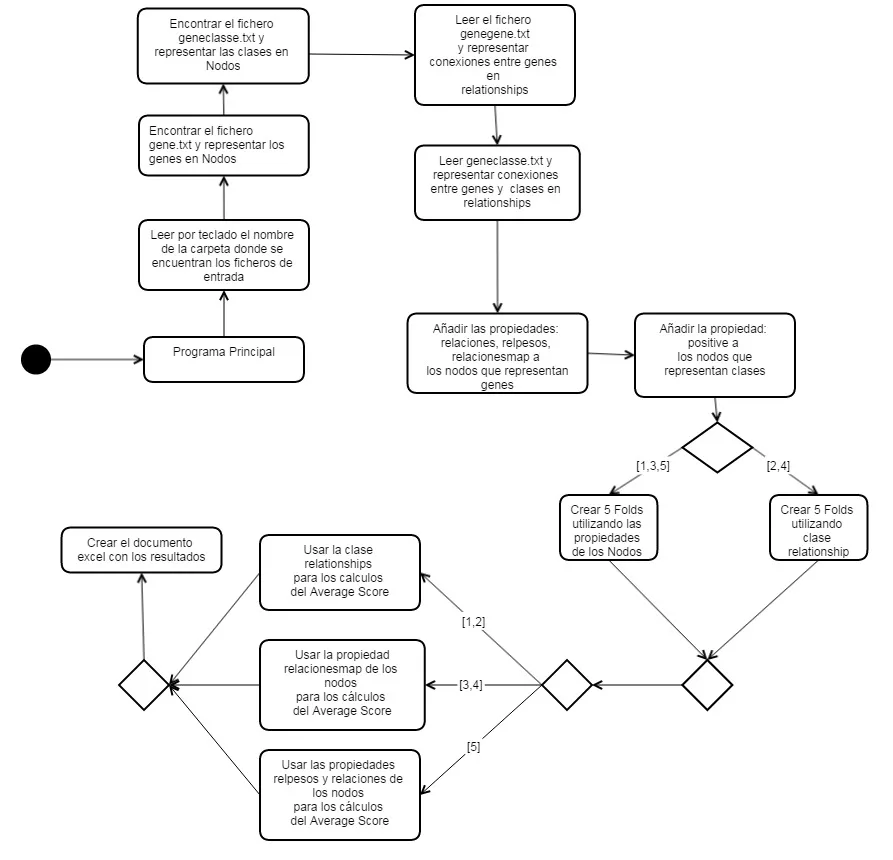
6.4. Implementación en Titan Cassandra
A continuación vamos a comentar la implementación del AvgScore para Titan utilizando Cassandra como base. Para realizar este experimento hemos utilizado la versión 0.4.0 de Titan para la gestión de grafos y 1.2.2 de Cassandra para almacenar la información, ambas obtenidas del sitio oficial de Titan. En los siguientes apartados explicaremos cómo hemos utilizado los diferentes mecanismos que nos proporciona Titan para resolver los algoritmos expuestos anteriormente. Los subapartados que nos vamos a encontrar son los siguientes:- Diagrama de clases
- Representación de la estructura de la base de datos
- Utilización del método validación cruzada.
- Implementación de la función Score.
- Diagrama de actividades.
6.4.1 Diagrama de clases
6.4.2 Creación de la estructura
Para crear una base de datos de Titan tenemos que diseñar su configuración a partir de las características del sistema que vamos a utilizar, es decir, un sistema no distribuido, en local y con índices de estructura Lucene que aprovechan estas características. Antes de continuar con la creación de los genes y sus conexiones tenemos que definir todos los índices que utilizaremos para aumentar la eficiencia de nuestro algoritmo, ya que el sistema necesita primero crear los índices para después rellenarlos con los vértices que contienen la información de forma automática. Con la estructura finalizada, tendremos que implementar los métodos que se encargará de introducir los genes, las clases genéticas y los arcos que los relacionan entre sí. Para hacer esta tarea hemos implementado los algoritmos expuestos en la sección 6.2.2 pero adoptando el código para este sistema y sus necesidades. En este sentido tenemos que recordar el sección 3.3 del proyecto donde hemos presentado las característica del sistema Titan y hemos explicado como no soporta eficientemente grafos con una cantidad muy elevada de conexiones. Sabiendo este hecho y para intentar obtener la mejor implementación posible de nuestros algoritmos hemos creado dos estructuras muy diferentes, una primera que crea las conexiones básicas para realizar consultas mediante arcos y otra en la que introduce esta información en las propiedades de los genes almacenados en la base de datos. Nuestra intención es comparar en los resultados estas dos implementaciones para ver cuál obtiene los mejores resultados.Versión con Vertex y Edge
Para hacer este experimento lo primero que debemos recordar es que Titan es un sistema que gestiona objetos llamados Vertex y Edges. Estas clases se corresponden a los vértices y los arcos de un grafo y las vamos a utilizar para representar los genes y sus conexiones respectivamente. Para ellos crearemos genes a través de los métodos de la clase Vertex con las siguientes propiedades:- nameGen: almacena el nombre del gen.
- type: indicará que el Vertex es un vertex
- nameClass: nombre de las clases.
- type: indicará que el Vertex es una clase.
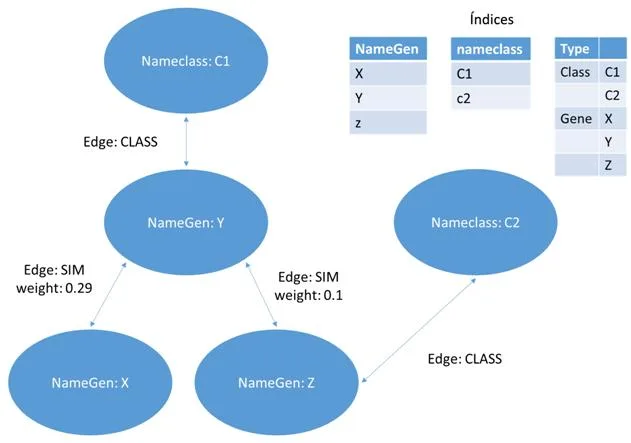
Versión con Vertex
La otra implementación posible es almacenar toda la información necesaria para los cálculos en los vértices. Aparte de las propiedades comentadas en la implementación anterior, al no existir la posibilidad de utilizar arcos para encontrar sus conexiones, tendremos que crear más propiedades en el Vertex que representa el gen para almacenar esta nueva información. Con esa idea hemos añadido las siguientes propiedades:- : contendrá la división de 1 entre la raíz cuadrada del valor de la suma de todas las relaciones.
- relaciones: array con todos los genes que tiene una conexión de similitud.
- pesosrelaciones: array con el valor numérico de la similitud. Existe una correlación entre este array y la propiedad relaciones.
- pesosrelacionesdjj: array con el valor numérico de Djj. Existe una correlación entre este array y la propiedad relaciones.
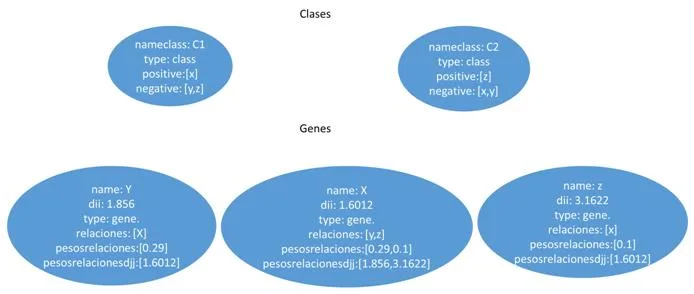
- create(String,String):El primer parámetro que recibimos contiene el nombre del directorio donde crear nuestra base de datos y el segundo donde almacenar los tiempos de nuestro programa. Dentro de este método está todos los procesos para la creación de nuestra estructura.
- setD(): este método recorre todos los genes de la base de datos para calcular el valor de que posee el gen analizado. Una vez que ha sido calculado lo almacena en el Vertex como una propiedad llamada “ ”. Para realizar esta tarea simplemente obtenemos el array con todos los valores de similitud llamado “pesosrelaciones” y realizamos los cálculos matemáticos pertinentes.
6.4.3 Validación cruzada
Las modificaciones que permiten adaptar este algoritmo (explicado en el apartado 6.2.2) a las bases de datos de Titan empiezan por modificar en la clase Fold los arrays positiveGenFold e negativeGenFold para gestionar Vertex, las clases que van a representar nuestros genes. La siguiente diferencia la vamos a encontrar en el método findGenesClassType(). Esta función nos proporciona como resultado todos los Vertex positivos y negativos a una clase y para este sistema de gestión de bases de datos hemos implementado dos métodos diferentes para realizar esta tarea: findGenesClassTypeEdge y findGenesClassTypefindGenesClassTypeEdge
Esta función se encargará de realizar una consulta en la base de datos para buscar todos los arcos del tipo “class” que salgan del Vertex proporcionado como argumento. La consulta simplemente tendrá que buscar todos los arcos salientes del tipo “class” que salgan del nodo de la clase analizada utilizando el getEdges().El resultado será devuelto mediante una variable del tipo iterable y por lo tanto con un simple for podemos recorrerla y almacenar en un array el contenido de la búsqueda, es decir todos los genes positivos respecto la clase. Con esa información podremos obtener la lista de los genes negativos y, por lo tanto, todos los genes que no estén en esta lista, serán negativos respecto a la clase y tendremos también el array con los vértices negativos.findGenesClassType
Esta función utiliza la implementación más local de la información. Como no existen las relaciones, para obtener los genes positivos a la clase solamente tenemos que buscar la propiedad positive que contiene esta información. Una vez obtenida es almacenada en una variable y devuelta para realizar los cálculos necesarios. El mayor problema que nos vamos a encontrar para crear esta función es que Titan cassandra no puede gestionar arrays de mucho tamaño. La solución por la que hemos optado es dividir los arrays en diferentes propiedades y por lo tanto tendremos que leer más de una propiedad para obtener los genes positivos y negativos a la clase.Otros métodos
Aparte de estos dos métodos la implementación de la validación cruzada se realizará en la clase Fold.6.4.4 Score
En la clase Scoring las modificaciones también están relacionadas con el sistema de consultas de Titan. Hemos implementado métodos especiales para conseguir el correcto funcionamiento de la función Score con la intención de adaptar todas las consultas al sistema Titan utilizado. Los métodos utilizados más importantes en esta clase son:- AvgScore(): Esta función realiza los cálculos matemáticos a partir de las propiedades que hemos almacenado en los Vertex. A partir de las etiquetas “ ” , ”pesosrelaciones” y “pesosrelacionesdjj” podremos obtener el valor de , el peso de similitud y los valores Djj respectivamente.
- AvgScore(): Esta función utiliza la clase Edge para obtener la información necesaria para realizar el Average Score. Mediante la función gedEdge podremos recorrer todas las conexiones del gen analizado para obtener los valores de similitud y de los vertex que necesitamos o, por otra parte, AvgScoreVertex().
6.4.5 Modelo de ejecución/navegación
Una vez descritos los posibles casos de uso que puede ejecutar el usuario junto con sus flujos de eventos vamos a describir la navegación que se puede realizar en la aplicación.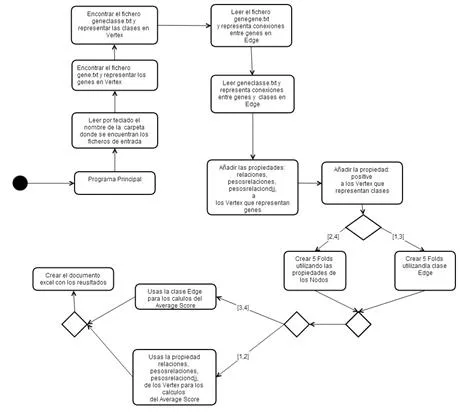
6.5. MongoDb
En los siguientes apartados explicaremos cómo hemos utilizado los diferentes mecanismos que nos proporciona MongoDb para resolver los algoritmos expuestos anteriormente. Los subapartados que nos vamos a encontrar son los siguientes:- Diagrama de clases
- Representación de la estructura de la base de datos
- Utilización del método validación cruzada.
- Implementación de la función Score.
- Diagrama de actividades.
6.5.1 Diagrama de clase
6.5.2 Representación de la estructura
El siguiente punto del proyecto es la implementación del algoritmo para MongoDb. Para realizar este experimento hemos utilizado la versión 2.11.3 de MongoDb obtenida del sitio oficial. En el próximo apartado explicaremos cómo hemos utilizado los diferentes mecanismos que nos proporciona este sistema para resolver los algoritmos expuestos anteriormente. Empezaremos primero con la creación de la base de datos y como representa la información almacenada para después implementar la función del Score, utilizando siempre como método de verificación el algoritmo validación cruzada.Estructura
Como habíamos expuesto en el apartado 3.3, MongoDb tiene una estructura orientada a documentos del tipo Json y, por lo tanto, tendremos que empezar creando los documentos que posteriormente vamos a introducir en la base de datos. Los documentos que representen a los genes los almacenaremos en la colección genes y tendrán siempre la siguiente propiedades clave valor:- _id: Campo hexadecimal utilizado como identificador del documento. Utilizaremos las id proporcionadas por el sistemas, explicadas en el apartado 2.
- name: Nombre del gen analizado.
- clases: Array donde almacenamos las clases en las que pertenece el gen representado.
- : valor numérico que representa este concepto matemático.
- rel : array donde almacenamos el valor de los genes con los que tiene relación.
- inforel:array con los valores de similitud de cada relación
- rel: array con el valor de la similitud que existe con los genes que está relacionado
- relname: array con los nombres de los genes que posee relación.
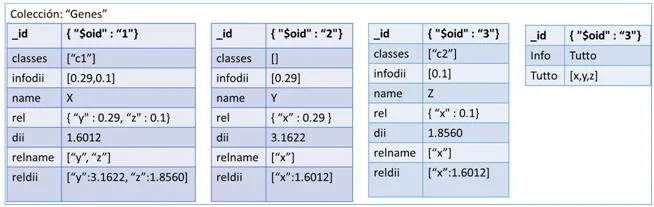
- _id: Campo hexadecimal utilizado como identificador del documento. Utilizaremos las id proporcionadas por el sistemas, explicadas en el apartado de Mongodb 3.2.
- Nombre: Nombre de la clase analizada.
- Gene: Array con los nombre de los genes que pertenecen a la clase
- Genenoclass”: Array con los nombre de los genes que no pertenecen a la clase
- "tuttoclass" : que contendrá "infclass" almacenado.
- infclass”: Array con los nombre de las clases almacenadas en el sistema.
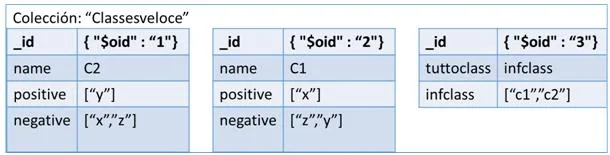
- id: Campo hexadecimal utilizado como identificador del documento. Utilizaremos las id proporcionadas por el sistemas.
- Nombre: Nombre de la conexión genética analizada. Estará formada por el nombre de los dos genes entre los que hay una relación, primero el saliente y después el entrante. Hemos optado por crear las relaciones en ambos sentidos, es decir si existe una conexión entre X e Y hemos creado dos documentos diferentes, uno llamado XY y otro YX.
- Weight: Valor numérico que contiene el valor de similitud de la conexión genética
- : Valor numérico que contiene que correspondería a este campo matemático
- Djj: Valor numérico que contiene que correspondería a este campo matemático

6.5.3 Validación cruzada
Las modificaciones que permiten adaptar este algoritmo (explicado en el apartado 6.2.2) a las bases de datos de MongoDb empiezan modificando los arrays positiveGenFold e negativeGenFold para gestionar String. El método findNodesClassType() ya no realizará búsquedas en vértices de grafos . Esta función se encargará de realizar consultas en la base de datos para buscar los documentos que poseen propiedades otorgados por parámetros. Con el lenguaje nativo de MongoDb, simplemente buscaremos y obtendremos de la colección de clases el documento que representa a la clase analizada. De este documento le indicaremos que queremos obtener obtener las propiedades gene y genenoclass para devolver la lista de los genes positivos y negativos a esa clase. Aparte de estos dos métodos la implementación de la validación cruzada se realizará en la clase Fold.6.5.4 Score
En la clase Scoring las modificaciones están relacionadas con el sistema de consultas de MongoDB y sus diferentes implementaciones. Primero de todo hemos implementado 3 métodos especiales para conseguir el correcto funcionamiento de la función Score que son:- AvgScore(): Para utilizar este método en la implementación las modificaciones que deberemos realizar estarán relacionadas con la búsqueda de documentos y extracción de sus propiedades. El primer paso es localizar en la colección de documentos de genes el documento que representa nuestro gen. Para ello simplemente tendremos que comprobar que el campo “name” de nuestro documento coincide con el nombre de Gen. Una vez encontrado tendremos que extraer los campos “ ”, “rel” y “info ” y a partir de ellos extraer toda la información que nos proporcionan y realizar las operaciones matemáticas necesarias.
- AvgScoreVersionRelaciones() Este método utiliza la colección relaciones para obtener la información de los genes conectados entre sí. Para obtenerla tenemos que buscar la relación y extraer la información, de un tamaño menos que el AvgScore()o el AvgScoreHashMap();
- AvgScoreVersionHashMap():En esta implementación pasamos de utilizar estructuras del tipo arrayList para almacenar la información de las relaciones para utilizar HashMap. De este modo las consultas devuelven menos información y puede ser gestionada más eficientemente. Para llevar a cabo esta idea utilizaremos la información almacenada en la propiedad "rel".
6.5.5 Modelo de ejecución/navegación
Una vez descritos los posibles casos de uso que puede ejecutar el usuario junto con sus flujos de eventos vamos a describir la navegación que se puede realizar en la aplicación.
6.6. Implementación en Cassandra
El siguiente punto del proyecto es la implementación del algoritmo para Cassandra. Para realizar este experimento hemos utilizado la versión 2.0.6 obtenida del sitio oficial. En los siguientes apartados explicaremos cómo hemos utilizado los diferentes mecanismos proporcionados por Cassandra para resolver la función Score basada en Kernels. Los subapartados que nos vamos a encontrar son los siguientes:- Diagrama de clases
- Representación de la estructura de la base de datos
- Utilización del método validación cruzada.
- Implementación de la función Score.
- Diagrama de actividades.
6.6.1 Diagrama de clase
6.6.2 Representación de la estructura
Como habíamos expuesto en la sección 2.5, Cassandra tiene una estructura orientada a columnas y, por lo tanto, empezaremos definiendo como son estas estructura que posteriormente vamos a introducir en la base de datos. Las dividiremos en tres apartados según representen a genes, clases o relaciones. Las column que representen a los genes los almacenaremos en la ColumnFamily llamada Genes y tendrán siempre los siguientes campos clave valor:- name: Nombre del gen analizado.Esta propiedad es la encargada de identificar la columna.
- clases: Array donde almacenamos las clases en las que pertenece el gen representado.
- : valor númerico que representa este concepto matemático.
- rel : array donde almacenamos el valor matemático de los genes con los que tiene relación.
- inforel:array con los valores de similitud de cada relación
- rel: array con el valor de la similitud que existe con los genes que está relacionado
- relname: array con los nombres de los genes que posee relación.
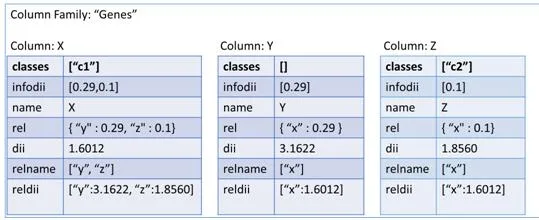
- Nombre: Nombre de la clase analizada.
- Gene: Array con los nombre de los genes que pertenecen a la clase
- Genenoclass: Array con los nombre de los genes que no pertenecen a la clase

- Nombre: Nombre de la conexión genética analizada. Estará formada por el nombre de los dos genes entre los que hay una relación, primero el saliente y después el entrante. Hemos optado por crear las relaciones en ambos sentidos, es decir si existe una conexión entre X e Y hemos creado dos documentos diferentes, uno llamado XY y otro YX. Esta propiedad es la encargada de identificar la columna.
- Weight: Valor numérico que contiene el valor de similitud de la conexión genética
- : Valor numérico que contiene que correspondería a este campo matemático
- Djj: Valor numérico que contiene que correspondería a este campo matemático
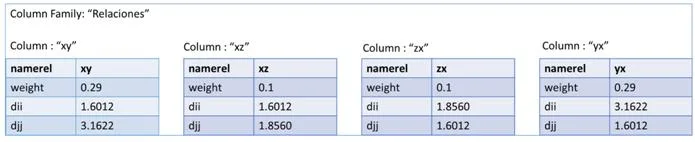
6.6.3 Validación cruzada
Las modificaciones que permiten adaptar este algoritmo (explicado en el apartado 6.2.3) a las bases de datos de Cassandra son muy parecidos a los de MongoDb, como por ejemplo que los arrays positiveGenFold e negativeGenFold van a gestionar String. El método findNodesClassType() Esta función se encargará de realizar consultas en la base de datos la columna con el nombre dado por parámetros. Con el CQL, simplemente buscaremos y obtendremos la columna que representa a la clase analizada. De esta columna le indicaremos que queremos obtener las filas que corresponden a gene y genenoclass para devolver el conjunto de los genes positivos y negativos a esa clase. Aparte de estos dos métodos la implementación de la validación cruzada se realizará en la clase Fold.6.6.4 Score
En la clase Scoring las modificaciones están relacionadas con el sistema de consultas de Cassandra y sus diferentes implementaciones . Primero de todo hemos implementado 3 métodos especiales para conseguir el correcto funcionamiento de la función Score que son:- AvgScore():El primer paso es localizar la columna que representa nuestro gen. Para ello simplemente tendremos que buscar la columna identificada con el nombre del gen. Una vez encontrado tendremos que extraer los campos “ ”, “rel” y “info ” y a partir de ellos extraer toda la información que nos proporcionan y realizar las operaciones matemáticas necesarias.
- AvgScoreVersionRelaciones() A partir de las columnas de la Column Family relaciones para obtener la información de los genes conectados entre sí. Para obtenerla tenemos que buscar la relación y extraer la información, de un tamaño menor que el AvgScore()o el AvgScoreHashMap();
- AvgScoreVersionHashMap():En esta implementación pasamos de utilizar estructuras del tipo arrayList para almacenar la información de las relaciones para utilizar HashMap. De este modo las consultas devuelven menos información y puede ser gestionada más eficientemente. Para llevar a cabo esta idea utilizaremos la información almacenada en la propiedad "rel".
6.6.5 Modelo de ejecución/navegación
Una vez descritos los posibles casos de uso que puede ejecutar el usuario junto con sus flujos de eventos vamos a describir la navegación que se puede realizar en la aplicación.
6.7. Verificación de resultados
Para verificar todos los resultados obtenidos en las implementaciones realizadas, el profesor Marco Messeti nos ha proporcionado los resultados obtenidos por un algoritmo escrito en R para une serie de ejemplos. La siguiente tabla nos muestra los resultados para el sample1 una base de datos con 10 genes y dos clases.
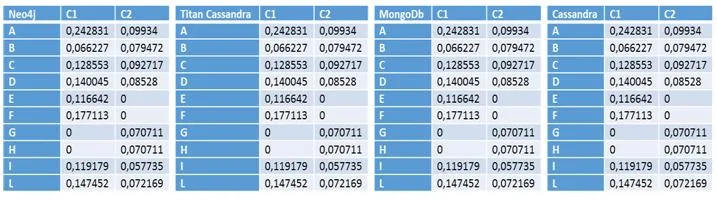
6.8. Resultados
Una vez verificados todos los resultados en todos los sistemas y en todas las posibles implementaciones, hemos empezado con el siguiente punto de nuestro proyecto, comparar todos los programas y sus implementaciones para analizar cuales poseen rendimientos mejores o una mayor eficiencia en determinadas circunstancias. Como hemos comentado ya, el mayor interés de nuestro proyecto es el análisis del rendimiento para la gestión de diferentes bases de datos con cantidades de información muy elevadas y, por lo tanto, no nos va importar el tiempo de creación de nuestro sistema o el tiempo que vamos a invertir devolviendo los resultados. El mayor interés lo vamos a encontrar en el tiempo que necesita una base de datos para responder las consultas que le enviemos. Con esta idea, hemos analizado todas las clases de cada programa y hemos observado que todas las consultas al sistema son realizadas por dos métodos: rankingAvg() y Fold(). Por lo tanto, hemos añadido a estos métodos para medir el tiempo y analizar el coste temporal de realizar estas consultas como muestra en la siguiente figura [6.29] :
- Muestras de control: Son 6 ejemplos de bases de datos que han sido proporcionadas por el profesor Marco Mesiti. No siguen ningún patrón de tamaño y sirven para realizar comprobaciones de tamaño.
6.30 Muestras de verificación - Muestras con gran cantidada de genes y clases: Son 5 bases de datos genéticas con una gran cantidad de genes y muy pocas conexiones entre ellos y las clases. (cambiar el nombre de las muestras por el correcto)
6.31 Muestras con gran cantidad de Vertex - Muestras con gran cantidad de conexiones: Son 5 bases de datos genéticas con una gran cantidad de conexiones entre genes y una cantidad menor de vértices.
6.32 Muestras con gran cantidad de conexiones
6.8.1 Resultados Neo4J.
cprovechando la estructura que hemos generado con Neo4J , hemos realizado 5 ejecuciones diferentes en nuestro sistema aprovechando las diferentes posibilidades que nos ofrecían y explicaremos a continuación.- Implementación 1 Utilizamos los métodos findNodesClassTypelocal() y AvgScore() fusionamos dos estructuras diferentes. Utilizaremos por una parte la propiedad positive de los nodos que representan las clases para generar los folds mientras que para calcular el Average Score utilizaremos las clases relationships.
- Implementación 2 Utilizamos las métodos findNodesClassType() y AvgScore() . Estos métodos utilizan la clase Relationships y Node para realizar las consultas.
- Implementación 3 Utilizamos los métodos findNodesClassTypelocal() y AvgScoreVersiónLocal(). El primer método utiliza la propiedad positive de los nodos que representan las clases para generar los folds mientras que para calcular el Average Score utilizaremos la propiedad relacionesmap que tiene almacenados los vertex con los que está relacionado y el valor de similitud en forma de HashMap.
- Implementación 4 Utilizamos las métodos findNodesClassType() y AvgScoreVersiónmap() fusionamos otra vez dos estructuras diferentes. Utilizaremos por una parte la clase relationships para generar los folds mientras que para calcular el Average Score utilizaremos la propiedad relacionesmap.
- Implementación 5 Esta última implementación es un poco especial. Hemos utilizado la implementación findNodesClassTypelocal() ya explicada en los casos anteriores y además ahora hemos introducido dos propiedades “relname” y “relpesos” donde hay almacenados arrays con los nombre de los genes que están conectados y los valores de similitud.
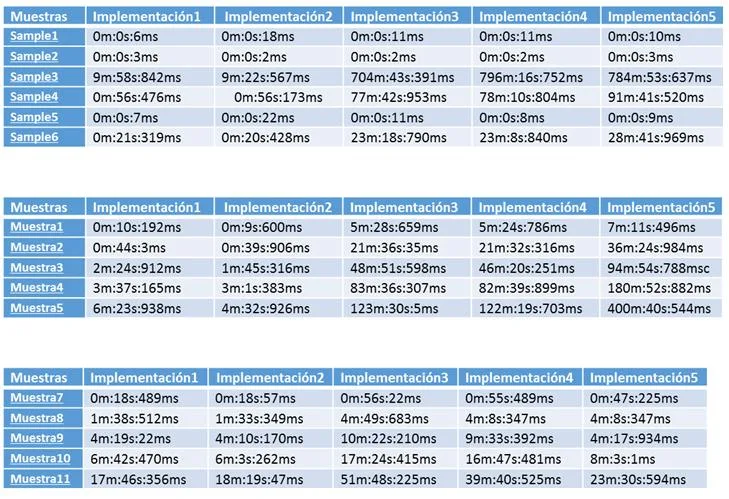
Cambios en la implementación no previstos.
El primer problema que nos hemos encontrado es que las propiedades en Neo4J sólo pueden almacenar información en String. Es decir que para todas los cosas donde hemos implementado una versión basada en estas estructuras, si queríamos almacenar algún dato en formato Array o HashMap teníamos que transformarlo en String y después hacer una conversión cuando realizamos la consulta mediante métodos como split o Replece de la clase String. Hemos desestimado después de las primeras pruebas con bases de datos de grandes dimensiones la opción de utilizar una propiedad “negative” en los nodos que generan las clases. Este hecho se ha producido después de que hayamos observado que la cantidad de memoria utilizada era demasiado grande y ralentizaba bastante cuando generaba los folds. Hemos sustituido esta implementación por otra que simplemente recorre todos los Nodos a través de un índice y comprueba si están contenidos en la propiedad positive.6.8.2 Resultados MongoDb.
Aprovechando la estructura que hemos generado con MongoDb hemos realizado 3 ejecuciones diferentes en nuestro sistema aprovechando las diferentes posibilidades que nos ofrecían y explicaremos a continuación.- Implementación 1 Utilizamos el método getsimandrel() con la intención de realizar una única consulta nos devuelva una gran cantidad de información y no tengamos la necesidad de realizar más
- Implementación 2 Utilizamos las métodos getHASHrel() y getelement() para obtener la información necesaria más específicamente. Realizamos más consultas pero las transacciones de información son más pequeñas
- Implementación 3 Utilizamos la colección relaciones. Realizamos consultas mucho más rápidas pero manejamos menos información. El proceso de construcción de esta implementación y la cantidad de memoria que necesita el constructor es mucho más elevado.
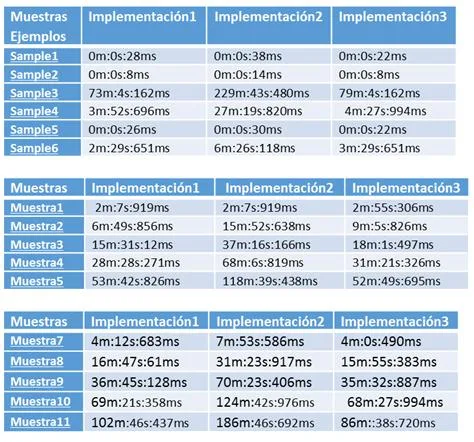
Cambios en la implementación no previstos.
Al momento de crear la base de datos, mongoDb no ha soportado la cantidad de insets enviados por el cliente. Para intentar no saturar el sistema hemos introducido un sleep cada 5.000 inserts o updates, consiguiendo nuestro objetivo de evitar saturar el sistema.6.8.3 Cassandra
Aprovechando la estructura que hemos generado con Cassandra explicado , hemos realizado 3 ejecuciones diferentes en nuestro sistema aprovechando las diferentes posibilidades que nos ofrecían y explicaremos a continuación.- Implementación 1 Utilizamos el método getsimandrel() con la intención de realizar una única consulta nos devuelva una gran cantidad de información y no tengamos la necesidad de realizar más
- Implementación 2 Utilizamos las métodos getHASHrel() y getelement() para obtener la información necesaria más específicamente. Realizamos más consultas pero las transacciones de información son más pequeñas
- Implementación 3 Utilizamos la colección relaciones. Realizamos consultas mucho más rápidas pero manejamos menos información. El proceso de construcción de esta implementación y la cantidad de memoria que necesita el constructor es mucho más elevado.
6.8.4 Resultados Titan Cassandra
Aprovechando la estructura que hemos generado con Neo4J, hemos realizado 4 ejecuciones diferentes en nuestro sistema aprovechando las diferentes posibilidades que nos ofrecían las estructuras creadas. Las implementaciones utilizadas son:- Implementación 1 Utilizamos el AvgScore() para aprovechar la información almacenada en las propiedades de los vértices y lo fusionamos con el método findGenesClassTypeEdge que utiliza la clase Edge para encontrar las clases de los elementos.
- Implementación 2 Utilizamos las métodos findGenesClassType() y AvgScore(), ambos basados en el uso de propiedades.
- Implementación 3 Utilizamos los métodos findGenesClassTypeEdge() y AvgScoreVersiónVertex(). Ambos basados en utilizar las clases Vertex y las clases Edge para obtener información y realizar cálculos.
- Implementación 4 Utilizamos los métodos findGenesClassType() y AvgScoreVersiónVertex(). El método para crear los folds se basa en las propiedades almacenadas en las clases mientras que el Average Score utiliza la clase Vertex y Edge para realizar los cálculos.
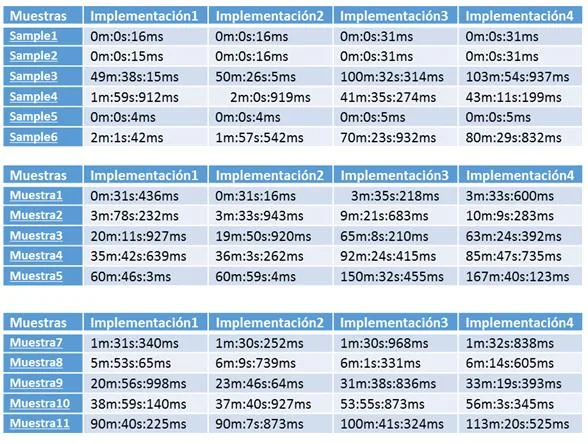
Cambios en la implementación no previstos
Hemos desestimado después de las primeras pruebas con bases de datos de grandes dimensiones la opción de utilizar una propiedad “negative” en los Vertex que generen las clases. Este hecho se ha producido después de que hayamos observado que la cantidad de memoria utilizada era demasiado grande y ralentizaba bastante cuando generaba los folds. Hemos sustituido esta implementación por otra que simplemente recorre todos los Vertex a través de un índice y comprueba si están contenidos en la propiedad positive.7. Conclusiones
7.1. Neo4J
Como podemos observar entre los diferentes diseños de implementación realizados en Neo4J, su mejor rendimiento se ha producido en la implementación más orientada a grafos (en comparación a otras más orientadas a las propiedades claves-valor). Estos resultados los ha repetido en todas las pruebas realizada con diferentes tamaños y cantidades de vértices .Las observaciones posteriores han reafirmado la eficiencia más elevada de una implementación utilizando el método getRelationships para aquellas bases de datos que tengan más relaciones. Esta causa es debida a una indexación optimizada para todas las conexiones entre nodos. Dado un nodo cualquiera, podemos obtener de forma muy veloz todos los nodos relacionados a través de sus conexiones. Respecto a las versión más locales, cabe decir que Neo4J solo permite almacenar datos del tipo String o tipo Double en sus propiedades. Este hecho ralentiza mucho las versiones que utilicen propiedades de forma abundante (3,4,5), ya que, aunque el tiempo necesario para consultar una propiedad en nodo Nodo no es muy elevado, el hecho de no poder almacenar pares clave-valor con valores tipo lista o hasmap obliga al analista a leer y tratar esta información para realizar cálculos con ellas. Esto significa que debes a partir de una fila de texto generar la estructura que vas a utilizar utilizando funciones como split o Replece con alto coste temporal. Para ver mejor las diferencia entre las implementaciones observa la figura [7.36] :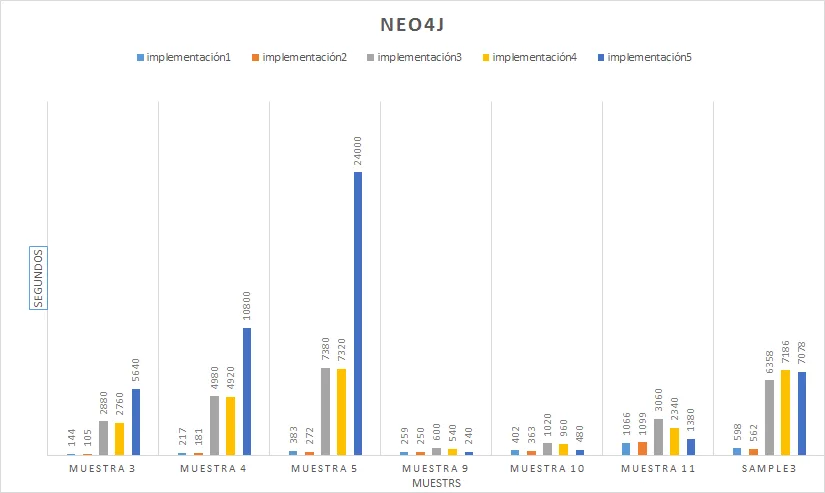
7.2 Titan Cassandra
Como podemos observar entre los diferentes diseños de implementación realizados en Titan Cassandra, su mejor rendimiento se ha producido en la implementación más local, es decir, aquella que utiliza las propiedades para obtener la información en las consultas. Estos resultados los ha repetido en todas las pruebas realizadas con diferentes tamaños y cantidades de vértices. En la documentación de Titan hallada en la web ya advertía sobre la ineficacia del sistema para grafos con muchas conexiones. Como no especificaba una cantidad exacta de conexiones a partir de los cuales el rendimiento empiece a escasear, hemos intentando probar con grafos con pocas conexiones en nuestros ejemplos pero el rendimiento obtenido ha dejado mucho que desear. A pesar de ser un sistema muy orientado a la distribución en diferentes computadores, hemos podido observar como la integración entre Cassandra y Titan para crear un gestor híbrido de bases de datos orientada a grafos ha sido aproximadamente 5 veces más lento en su versión más óptima que la utilizada por un gestor de grafos puro como Neo4J. También otro aspecto a comentar, aunque nuestras pruebas no miden este valor, son los tiempos de creación de una base de datos Titan Cassandra. Para crear una nueva base de datos como el Sample3 y la muestra11 ha llegado a sobrepasar las 24 horas, mientras que en Neo4J por ejemplo el caso más costoso ha sido creado en 50 minutos como máximo. Todos los resultados obtenidos se pueden compararse en la siguiente figura [7.37] :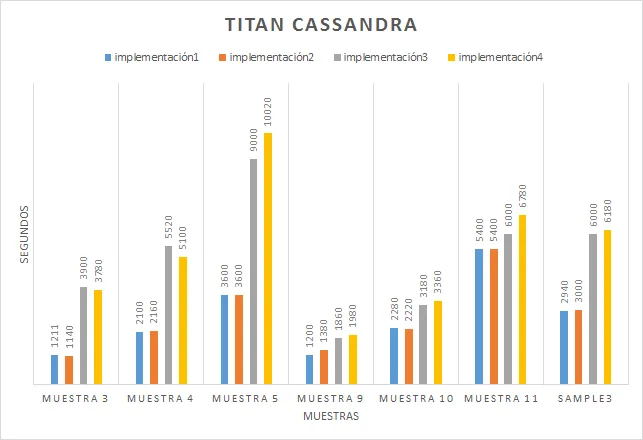
7.3. MongoDb
Como podemos observar entre los diferentes diseños de implementación realizados en MongoDb, su mejor rendimiento se ha producido en la implementación tanto utilizando los documentos de la colección relaciones como a través de la colección genes y sus propiedades. La implementación 2 ha utilizado métodos de la librería String para elaborar el algoritmo y, usar sus métodos métodos, genera una pérdida muy grande de prestaciones. Aparte de eso, en la documentación también advierten de una correlación entre la memoria ram disponible del ordenador con la velocidad de acceso a los documentos. Esto quiere decir que si invertimos mas ram en el computador funcionaria más rápidamente la implementación 3 que la 1, es decir, encontraríamos antes un documento en la colección relaciones y podríamos extraer la información en una consulta muy pequeñas que si lo comparamos en la implementación 1 con menos documentos donde buscar pero consultas más grandes y que necesitan más tiempo. Un pequeño detalle, es que la implementación 3, cuando ha tenido menos carga de documentos, es decir, en las muestras 11,10 y 9 ha sido más rápido que la implementación 1 con mucha más cantidad de vértices.Dicho todo esto los resultados los hemos expuesto en la siguiente figura.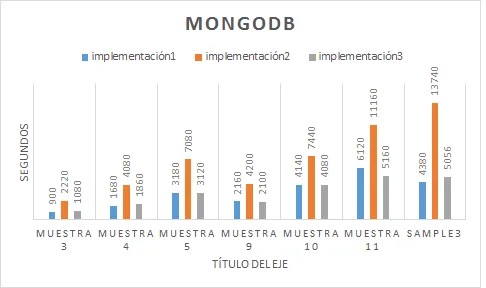
7.4 Cassandra
Respecto a Cassandra la verdad no ha cumplido las expectativas. Teóricamente una base de datos NoSQL está diseñada para ser soportada por ordenadores sin grandes recursos hardware pero un ordenador personal no ha conseguido soportar este sistema en condiciones. Según las investigaciones realizadas, cassandra (a parte de necesitar una cantidad de recursos muy elevada) cuando se implementa en local actúa igual como si estuviera siendo un sistema distribuido, por lo tanto si para hacer un insert o realizar una consulta realiza todas las comprobaciones correspondientes a los sistemas distribuidos en un ordenador con recursos muy limitados generan tiempos de lecturas y tiempos de escritura demasiados elevados. Por lo tanto, este sistema queda totalmente desestimado.7.5. Resultados en común
Al comparar todos los sistemas en los cuales hemos obtenidos resultados analizables, observamos que los gestores de bases de datos orientados a grafos son los sistemas que han proporcionado mejores prestaciones para realizar los cálculos más rápidamente. Por ejemplo con Neo4J hemos obtenido unos resultados entre 6 y 7 veces más veloces que en MongoDb. La razón de estos hechos, puede ser debida a la utilización de un ordenador personal con una memoria ram no muy elevada. Buscando información acerca de este aspecto en la documentación de MongoDb hemos comprobado comentarios de los usuarios están exponiendo problemas de lentitud en las consultas para servidores con una memoria ram no muy elevada. Exponen que para utilizar la indexación de los documentos y encontrar la información necesaria es necesario una cantidad de ram elevada para funcionar a un nivel veloz. Aparte de esto, el sistema está hecho más para bases de datos distribuidas, es decir, situaciones con escalabilidad horizontal donde se puede compartir recursos entre las diferentes máquinas. Realmente ha ocurrido un caso similar con Cassandra, pero mucho más extremo. Este sistema de gestión de base de datos está diseñado para utilizar y dividir la información en diversos nodos y, de este modo, poder aprovechar los recursos de cada una de las máquinas. Cuando hemos intentado implementar una versión local de esta base de datos, no hemos podido obtener información de forma muy optimizada y eficiente porque realizaba los mismos pasos analizados en la figura 2.11 para sistemas distribuidos utilizando una única máquina, es decir, realiza mucho trabajo innecesario. Este hecho ha impedido realizar consultas de forma masiva a la base de datos ya que los tiempos de espera eran demasiado elevados. Por último, comparar Neo4J y Titan Cassandra también nos resulta muy útil. Titan es un gestor de grafo muy deficiente. En la documentación básica que hemos analizado en el punto 2.5 ya hemos comentado el hecho que Titan no puede gestionar eficientemente los grafos con una cantidad muy elevada de aristas. La solución que hemos intentado implementar fue utilizar la clase Vertex fusionada con el motor de base de datos de Cassandra, una versión antigua adaptada para Titan que permite almacenar las propiedades de los nodos de forma rápida. Se considero que si implementabamos una versión más local, sin utilizar edges y almacenado la información de las aristas en los propoios Nodos podriamos llegar a obtener unos resultados del estilo de Neo4J. Pero no fue así. El rendimiento obtenido es de unas 5 o 6 veces más lento que Neo4J en los casos más grandes. Respecto a Neo4J cabe mencionar que de entre todas las versiones disponibles en el site, hemos buscado aquella que nos ofrecía más rendimiento en modo local ya que por ejemplo a partir de la versión 2.0 empieza a estar más diseñada para versiones distribuidas y por lo tanto no sería tan eficiente para nuestros experimentos. Otro punto importante a comentar como conclusión es la impotencias de no poder almacenar solamente doubles y strings en los Nodos. Si nuestro experimento necesitará almacenar otro tipo de información, deberíamos transformar la estructura a string y después realizar las conversiones que deseamos, con un coste computacional muy elevado. Por último presentamos una comparación de resultados, para ver claramente las diferencias de tiempo para cada implementación. Recordamos que en la sección 6.3 de este capítulo pueden encontrar el tamaño de las bases de datos utilizadas.